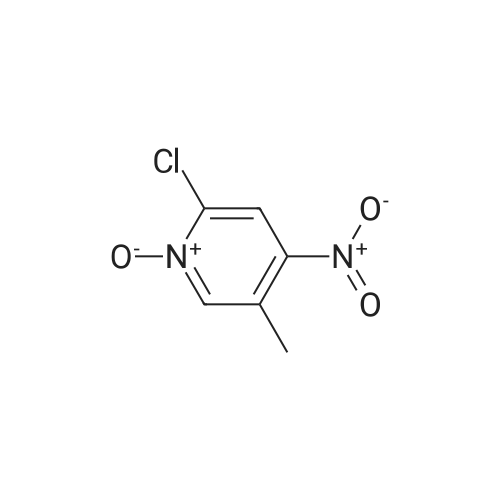
[ 14432-16-7 ] Synthesis Path-Downstream 1~90
- 1
-
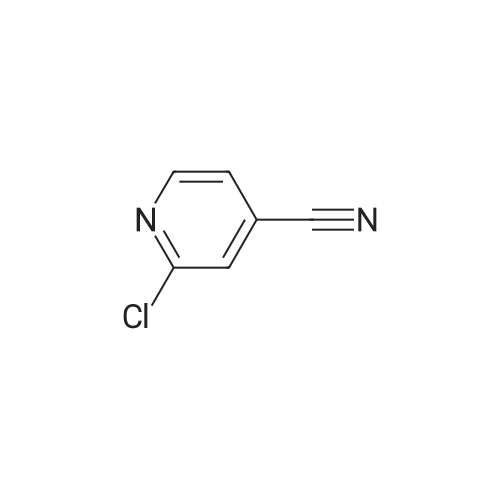
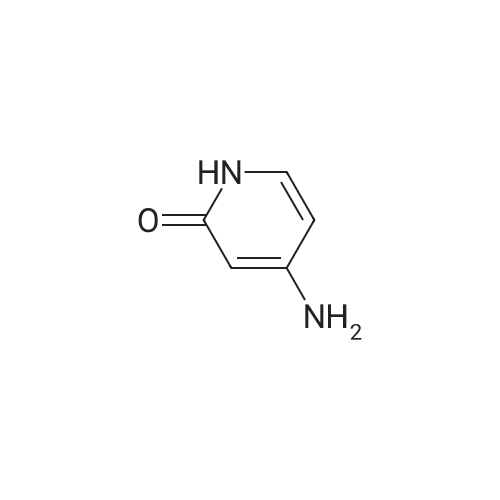
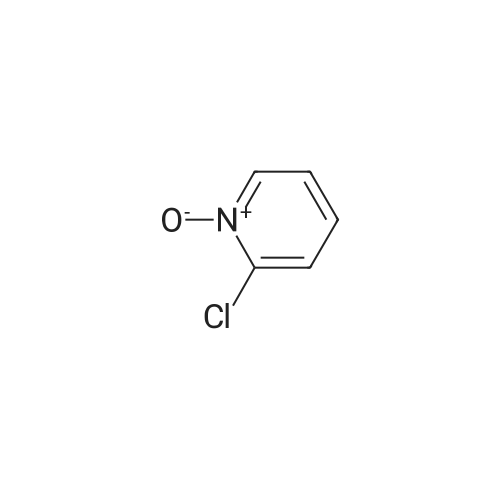 [ 2402-95-1 ]
[ 2402-95-1 ]

-
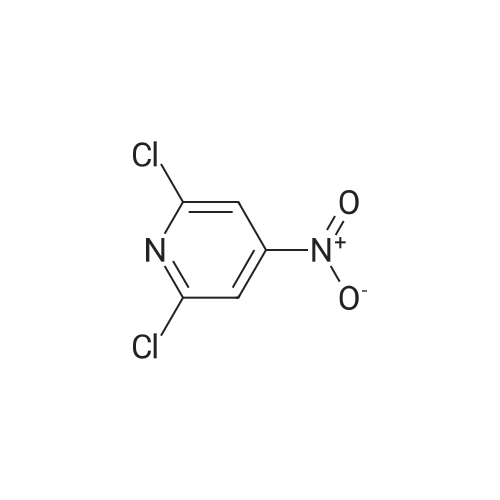
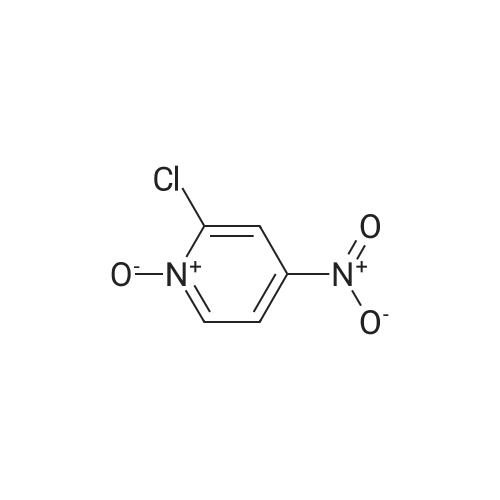 [ 14432-16-7 ]
[ 14432-16-7 ]
| Yield | Reaction Conditions | Operation in experiment |
| 93% |
nitration; |
|
| 78% |
With sulfuric acid; nitric acid at 90℃; for 4h; |
|
| 72% |
With sulfuric acid; nitric acid for 2h; |
|
| 65% |
With sulfuric acid; nitric acid at 0 - 100℃; |
1.2; 2.2; 3.2
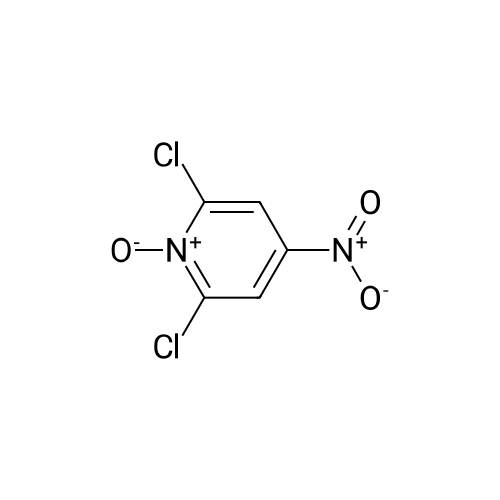
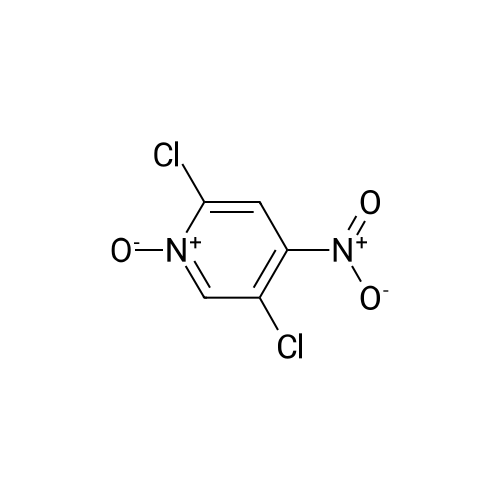
(2) was added 250ml three-neck flask in concentrated H2SO4(16ml), ice-water was cooled to 0 °C, was added dropwise concentrated nitric acid (10ml) formed mixed acid, at 0 °C, was added portionwise 2-chloropyridine N-oxide (6.4g, 0.05mol), the addition was complete 0-10 °C stirred for 20min, then heated to the reaction stirred for 100 °C, TLC to monitor the completion of the reaction, after the reaction was quenched with ice water, suction filtered to give 2-chloro-4-nitropyridine N-oxide, the molar yield was 65% |
| 65% |
With sulfuric acid; nitric acid at 0 - 100℃; |
1.2; 2.2; 3.2
(2) Add concentrated H2SO4(16ml) to a250ml three-necked bottle, cool to 0 ° C in ice water, add concentrated nitric acid (10ml) dropwise to form a mixed acid. (6.4g, 0.05mol), add 0-10 and stir for 20min, then increase the temperature to 100 and stir the reaction. TLC monitor until the reaction is complete. After the reaction is complete, quench with ice water and filter with suction to obtain 2-chloro-4- Nitropyridine nitrogen oxide, the molar yield is 65%; |
| 56% |
With nitric acid In concentrated H2 SO4; chloroform |
1.1 (1)
(1) 2-Chloro-4-nitropyridine-1-oxide 2-Chloro-pyridine-1-oxide (10 g) is cooled in an ice bath and treated with concentrated H2 SO4 (15 ml), followed by the dropwise addition of a mixture of concentrated H2 SO4 (15 ml) and fuming HNO3 (27 ml, s.g. 1.5) over a 70 minute period. The acidic solution is heated in a steam bath for 2.5 hours, then allowed to reach room temperature and poured onto ice water (600 ml), stirring being continued until all the ice has melted. The resultant solid is filtered off and dissolved in hot chloroform, the solution being dried and the solvent evaporated in vacuo to give a yellow solid. The aqueous filtrate obtained after the removal of the original solid is neutralised with saturated aqueous Na2 CO3 and extracted continuously with chloroform, the extract being dried and evaporated in vacuo to yield a yellow solid. The two yellow solids are combined and recrystallized from ethanol to give 2-chloro-4-nitro-pyridine-1-oxide as yellow crystals (7.46 g, 56%). |
|
With sulfuric acid; nitric acid |
|
|
With sulfuric acid; nitric acid |
|
|
With sulfuric acid; nitric acid at 90℃; for 2.5h; Yield given; |
|
|
With sulfuric acid; nitric acid |
D
;Reaction of 2-chloropyridine (CAS 109-09-1) with 3-chloroperoxybenzoic acid (CAS 937-14-4) gives compound 1 (Chem. Commun., 2000, 1577). Reaction of compound 1 with a mixture of nitric and sulfuric acids leads to compound 2 (J. Amer. Chem. Soc., 1959, 81, 2674). Reaction of compound 2 with sodium methoxide affords 2,4-dimethoxypyridine 1-oxide 3-2 (J. Chem. Soc., Perkin Trans 1, 1990, 1623). |
|
With sulfuric acid; nitric acid at 90℃; |
|

Reference:
[1]Alker, David; Ollis, W. David; Shahriari-Zavareh, Hooshang
[Journal of the Chemical Society. Perkin transactions I, 1990, # 6, p. 1623 - 1630]
[2]Schnekenburger; Riedel
[Archiv der Pharmazie, 1982, vol. 315, # 10, p. 825 - 831]
[3]Searls, Tim; McLaughlin, Larry W.
[Tetrahedron, 1999, vol. 55, # 41, p. 11985 - 11996]
[4]Current Patent Assignee: DANYANG NINGDA WEIFANG DETECTION TECH - CN109748865, 2019, A
Location in patent: Paragraph 0016; 0019; 0021; 0023; 0025; 0028; 0030
[5]Current Patent Assignee: CHANGZHOU CHUANYOU ENVIRONMENTAL PROTECTION TECH - CN111056999, 2020, A
Location in patent: Paragraph 0016; 0019; 0021; 0023; 0025; 0028; 0029
[6]Current Patent Assignee: BOSTON SCIENTIFIC CORP - US4587240, 1986, A
[7]Finger; Starr
[Journal of the American Chemical Society, 1959, vol. 81, p. 2674]
Brown
[Journal of the American Chemical Society, 1957, vol. 79, p. 3565]
Talik; Plazek
[Roczniki Chemii, 1955, vol. 29, p. 1019,1025][Chem.Abstr., 1956, p. 12045]
[8]Oszust; Talik; Pietraszko; Marchewka; Baran
[Journal of Molecular Structure, 1997, vol. 415, # 1-2, p. 53 - 63]
[9]Barraclough, Paul; Gillam, Janet; King, W. Richard; Nebbs, Malcolm S.; Vine, Susan J.
[Journal of Chemical Research, Miniprint, 1997, # 6, p. 1359 - 1376]
[10]Current Patent Assignee: BIOSAFE (unclear); PROCTER & GAMBLE CO - US2006/156481, 2006, A1
Location in patent: Page/Page column 9
[11]Lougiakis, Nikolaos; Gavriil, Efthymios-Spyridon; Kairis, Markelos; Sioupouli, Georgia; Lambrinidis, George; Benaki, Dimitra; Krypotou, Emilia; Mikros, Emmanuel; Marakos, Panagiotis; Pouli, Nicole; Diallinas, George
[Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 22, p. 5941 - 5952]
- 2
-
[ 14432-16-7 ]
-
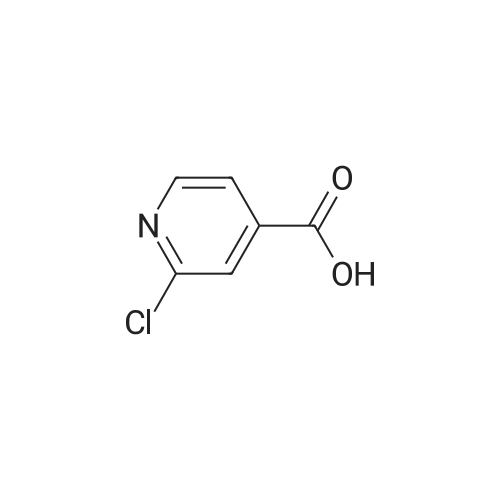
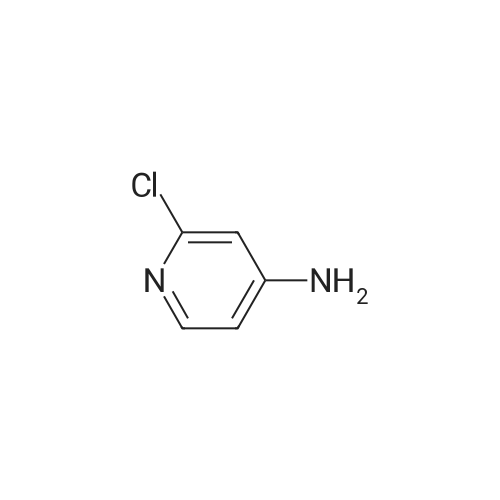 [ 14432-12-3 ]
[ 14432-12-3 ]
| Yield | Reaction Conditions | Operation in experiment |
| 95% |
With hydrogenchloride; iron In ethanol for 3h; Heating; |
|
| 90% |
With hydrogen In methanol |
|
| 85% |
With hydrogenchloride; iron In ethanol; water Reflux; |
1.3
(3) 2-chloro-4-nitropyridine N-oxide (7.9g, 0.05mol) in 500ml three-neck flask, iron powder (14g, 0.25mol), ethanol 150ml, water 50ml, 5ml of concentrated hydrochloric acid was heated to reflux , TLC to monitor the completion of the reaction, ethyl acetate was added until the reaction was complete 30ml, stirring was allowed to stand suction, delamination, dried over anhydrous sodium sulfate, and concentrated to give the product 2-chloro-4-aminopyridine, molar yield 85% |
| 85% |
With hydrogenchloride; iron In ethanol; water Reflux; |
1.3
(3) In a 500ml three-necked bottle, add 2-chloro-4-nitropyridine nitrogen oxide (7.9g, 0.05mol), iron powder (14g, 0.25mol), 150ml of ethanol, 50ml of water, and 5ml of concentrated hydrochloric acid to warm to reflux , TLC monitor until the reaction is complete. After the reaction is completed, add 30ml of ethyl acetate, stir and leave to filter with suction, separate the layers, dry over anhydrous sodium sulfate, and concentrate to obtain the product 2-chloro-4-aminopyridine, with a molar yield of 85%. |
|
With iron; acetic acid anschliessend mit Zink-Pulver unter Zusatz von wenig wss. HgCl2; |
|
|
In tetrahydrofuran for 0.25h; Ambient temperature; Yield given; |
|
|
With hydrogenchloride; iron In ethanol; water Reflux; |
|
Reference:
[1]Searls, Tim; McLaughlin, Larry W.
[Tetrahedron, 1999, vol. 55, # 41, p. 11985 - 11996]
[2]Barraclough, Paul; Gillam, Janet; King, W. Richard; Nebbs, Malcolm S.; Vine, Susan J.
[Journal of Chemical Research, Miniprint, 1997, # 6, p. 1359 - 1376]
[3]Current Patent Assignee: DANYANG NINGDA WEIFANG DETECTION TECH - CN109748865, 2019, A
Location in patent: Paragraph 0016; 0019; 0022
[4]Current Patent Assignee: CHANGZHOU CHUANYOU ENVIRONMENTAL PROTECTION TECH - CN111056999, 2020, A
Location in patent: Paragraph 0016; 0019; 0022
[5]Talik; Plazek
[Roczniki Chemii, 1955, vol. 29, p. 1019,1025][Chem.Abstr., 1956, p. 12045]
[6]Malinowski, Marek; Kaczmarek, Lukasz
[Journal fur praktische Chemie (Leipzig 1954), 1988, vol. 330, # 1, p. 154 - 158]
[7]Lougiakis, Nikolaos; Gavriil, Efthymios-Spyridon; Kairis, Markelos; Sioupouli, Georgia; Lambrinidis, George; Benaki, Dimitra; Krypotou, Emilia; Mikros, Emmanuel; Marakos, Panagiotis; Pouli, Nicole; Diallinas, George
[Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 22, p. 5941 - 5952]
- 3
-
[ 14432-16-7 ]
-
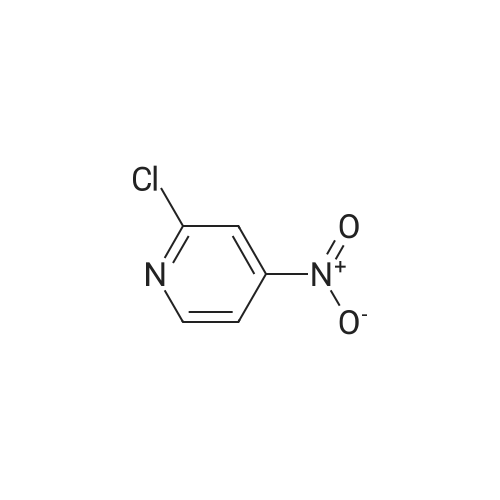 [ 23056-36-2 ]
[ 23056-36-2 ]
| Yield | Reaction Conditions | Operation in experiment |
| 88% |
With phosphorus trichloride In dichloromethaneu for 4h; Reflux; |
|
| 78% |
With phosphorus trichloride In chloroform at 20℃; Heating / reflux; |
17
Reference Example 17: 2-Chloro-4-nitro-pyridine. Phosphorus trichloride (4.2 mL, 48.7 mmol) was-added_to a solution of 2- chloro-4-nitro-pyridine-l -oxide (1.70 g, 9.74 mmol) in dry chloroform (25 mL) at r.t.The reaction mixture was then heated to reflux and maintained at this temperature overnight. The reaction was cooled to r.t. then poured onto ice, basified to between pH7-8 with saturated aq. sodium bicarbonate solution and extracted with chloroform (x 2).The combined organic phase was washed with water and brine, dried over sodium sulfate and concentrated. Drying under high vacuum afforded 2-chloro-4-nitro-pyridine(1.2 g, 78 %) as a solid. |
|
With ethyl acetate; phosphorus trichloride |
|
Reference:
[1]Jankowiak, Aleksandra; Kaszynski, Piotr
[Journal of Organic Chemistry, 2009, vol. 74, # 19, p. 7441 - 7448]
[2]Current Patent Assignee: F2G LTD; F2G BRITISH BODY CORPORATE - WO2008/62182, 2008, A1
Location in patent: Page/Page column 114
[3]Finger; Starr
[Journal of the American Chemical Society, 1959, vol. 81, p. 2674]
- 4
-
 [ 110-91-8 ]
[ 110-91-8 ]

-
[ 14432-16-7 ]
-
 [ 35981-62-5 ]
[ 35981-62-5 ]
| Yield | Reaction Conditions | Operation in experiment |
| 63% |
In ethanol at 20℃; for 3h; |
|
|
In ethanol |
|
Reference:
[1]Matulenko, Mark A.; Paight, Ernest S.; Frey, Robin R.; Gomtsyan, Arthur; DiDomenico Jr., Stanley; Jiang, Meiqun; Lee, Chih-Hung; Stewart, Andrew O.; Yu, Haixia; Kohlhaas, Kathy L.; Alexander, Karen M.; McGaraughty, Steve; Mikusa, Joseph; Marsh, Kennan C.; Muchmore, Steven W.; Jakob, Clarissa L.; Kowaluk, Elizabeth A.; Jarvis, Michael F.; Bhagwat, Shripad S.
[Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 4, p. 1586 - 1605]
[2]Sazonov,N.V. et al.
[Pharmaceutical Chemistry Journal, 1972, vol. 6, # 3, p. 146 - 149][Khimiko-Farmatsevticheskii Zhurnal, 1972, vol. 6, # 3, p. 18 - 21]
- 6
-
[ 14432-16-7 ]
-
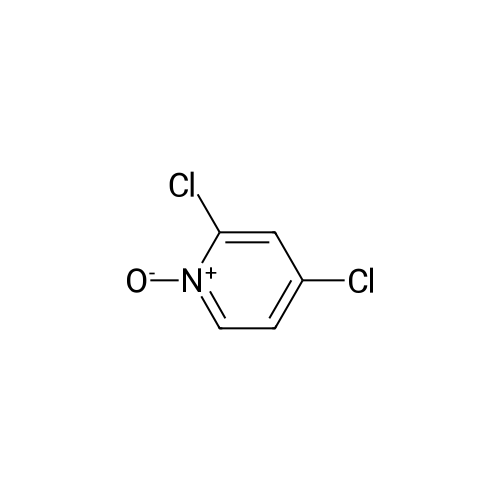 [ 13602-59-0 ]
[ 13602-59-0 ]
| Yield | Reaction Conditions | Operation in experiment |
| 94% |
With acetyl chloride at 70 - 75℃; for 0.416667h; |
|
| 80% |
With acetyl chloride for 0.666667h; |
|
Reference:
[1]Abramovitch, Rudolph A.; Deeb, Ali; Kishore, Dharma; Mpango, George B. W.; Shinkai, Ishiro
[Gazzetta Chimica Italiana, 1988, vol. 118, # 3, p. 167 - 172]
[2]Jankowiak, Aleksandra; Kaszynski, Piotr
[Journal of Organic Chemistry, 2009, vol. 74, # 19, p. 7441 - 7448]
- 8
-
[ 109-09-1 ]
-
[ 14432-16-7 ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: 91 percent / aq. H2O2 / trifluoroacetic acid / 4 h
2: 72 percent / aq. HNO3, H2SO4 / 2 h |
|
|
Multi-step reaction with 2 steps
1: 30percent aq. H2O2, AcOH / 48 h / 60 °C
2: HNO3, conc. H2SO4 / 2.5 h / 90 °C |
|
|
Multi-step reaction with 2 steps
1: H2O2 / acetic anhydride
2: HNO3, H2SO4 |
|
|
Multi-step reaction with 2 steps
1: 75 percent / aq. H2O2, glacial acetic acid / 12 h / 80 °C
2: 93 percent / nitration |
|
|
Multi-step reaction with 2 steps
1: peroxyacetic acid; acetic acid
2: concentrated sulfuric acid; nitric acid |
|
|
Multi-step reaction with 2 steps
1: 3-chloro-benzenecarboperoxoic acid / dichloromethane / 20 °C
2: nitric acid; sulfuric acid / 90 °C |
|
|
Multi-step reaction with 2 steps
1: 3-chloro-benzenecarboperoxoic acid / chloroform / 10 h / 25 °C
2: sulfuric acid; nitric acid / 0 - 100 °C |
|
Reference:
[1]Searls, Tim; McLaughlin, Larry W.
[Tetrahedron, 1999, vol. 55, # 41, p. 11985 - 11996]
[2]Barraclough, Paul; Gillam, Janet; King, W. Richard; Nebbs, Malcolm S.; Vine, Susan J.
[Journal of Chemical Research, Miniprint, 1997, # 6, p. 1359 - 1376]
[3]Oszust; Talik; Pietraszko; Marchewka; Baran
[Journal of Molecular Structure, 1997, vol. 415, # 1-2, p. 53 - 63]
[4]Alker, David; Ollis, W. David; Shahriari-Zavareh, Hooshang
[Journal of the Chemical Society. Perkin transactions I, 1990, # 6, p. 1623 - 1630]
[5]Finger; Starr
[Journal of the American Chemical Society, 1959, vol. 81, p. 2674]
Brown
[Journal of the American Chemical Society, 1957, vol. 79, p. 3565]
[6]Lougiakis, Nikolaos; Gavriil, Efthymios-Spyridon; Kairis, Markelos; Sioupouli, Georgia; Lambrinidis, George; Benaki, Dimitra; Krypotou, Emilia; Mikros, Emmanuel; Marakos, Panagiotis; Pouli, Nicole; Diallinas, George
[Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 22, p. 5941 - 5952]
[7]Current Patent Assignee: DANYANG NINGDA WEIFANG DETECTION TECH - CN109748865, 2019, A
Current Patent Assignee: CHANGZHOU CHUANYOU ENVIRONMENTAL PROTECTION TECH - CN111056999, 2020, A
- 10
-
[ 14432-16-7 ]
-
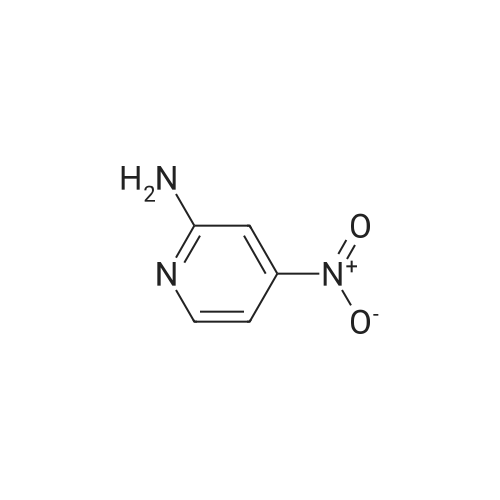 [ 4487-50-7 ]
[ 4487-50-7 ]
Reference:
[1]Journal of Molecular Structure,1997,vol. 415,p. 53 - 63
[2]Journal of Medicinal Chemistry,2011,vol. 54,p. 1836 - 1846
[3]Patent: WO2011/146591,2011,A1
[4]Patent: US2012/15942,2012,A1
[5]Patent: EP3305788,2018,A1
- 13
-
[ 14432-16-7 ]
-
[ 124-41-4 ]
-
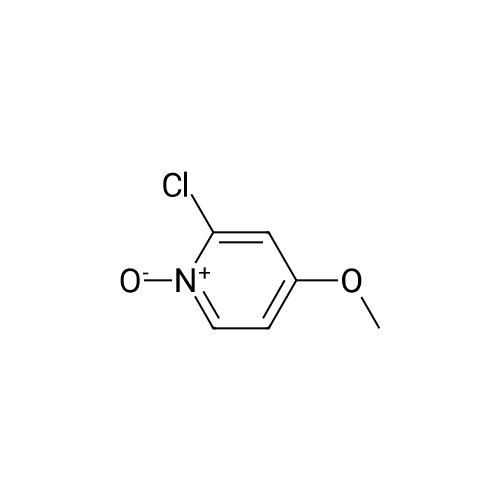 [ 38608-87-6 ]
[ 38608-87-6 ]
| Yield | Reaction Conditions | Operation in experiment |
| 94% |
With sulfuric acid; nitric acid In methanol at 20℃; for 48h; |
|
|
In methanol |
32.B
[242] Step B: 2-Chloro-4-methoxypyridine 1 -oxide:A solution of sodium methoxide, prepared by dissolving sodium (21 g) in dry MeOH (1000 ml), was quickly poured into a stirred solution of 2-chloro-4-nitropyridine 1 -oxide (151 g) in MeOH (1000 ml). Dissolution was complete within 5 minutes and the reaction was left stirring in a stoppered flask overnight. Partial concentration of the yellow solution produced a precipitate, which was filtered and washed with MeOH (2 x 100 mL). The filtrate and washings were evaporated to dryness, and the solid that remained was extracted with boiling dichloromethane to give (after filtration and concentration) a yellow-brown solid which was used in the next step without further purification. Yield= 95%. 1H NMR (400 MHz, CDCl3): δ= 8.256-8.237 (d, 1 H, J= 7.6 Hz), 7.026-7.018 (d, 1 H, J= 3.2 Hz), 6.804-6.777 (dd, 1 H, J= 3.6 & 3.2 Hz), 3.875 (s, 3 H). |
Reference:
[1]Current Patent Assignee: KUREHA CORP. - US6339045, 2002, B1
Location in patent: Referential example 2
[2]Current Patent Assignee: CHEMIZON BEIJING; KAIMEILONG BEIJING PHARMACEUTICAL TECHNOLOGY - WO2010/145197, 2010, A1
Location in patent: Page/Page column 80
- 14
-
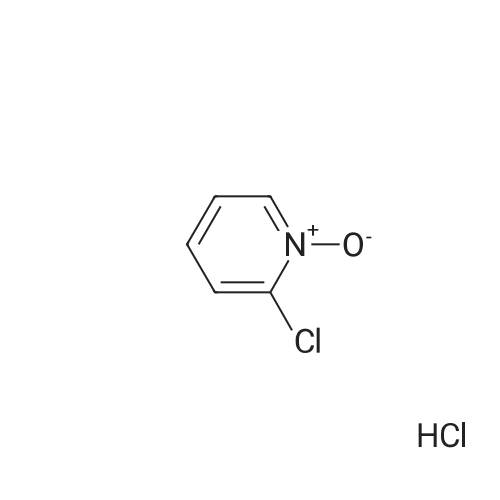 [ 20295-64-1 ]
[ 20295-64-1 ]
-
[ 14432-16-7 ]
| Yield | Reaction Conditions | Operation in experiment |
| 82.5% |
With sulfuric acid; nitric acid at 90 - 100℃; for 2.5h; |
|
| 37% |
With sodium hydroxide; nitric acid In sulfuric acid |
2.a a)
a) 2-Chloro-4-nitropyridine-N-oxide A solution of conc. H2SO4 (30 mL) and fuming HNO3 (54 mL) was added dropwise at 0° C. to a solution of 2-chloropyridine-N-oxide hydrochloride (15.2 g, 91.56 mmole) in conc. H2SO4 (30 mL). The reaction mixture was heated at 90° C. for 1 hr, then was cooled to RT and poured onto ice (500 g). The reaction mixture was kept at RT overnight, then was cooled in an ice bath, and 50% NaOH was added slowly to give a precipitate. This was collected and dried to give the title compound (5.88 g, 37%) as a pale yellow solid: 1H NMR (400 MHz, CDCl3) δ8.42-8.37 (m, 2 H), 8.06-8.04 (m, 1 H). |
|
With sulfuric acid; nitric acid In (2S)-N-methyl-1-phenylpropan-2-amine hydrate |
P.10.1 Production of 4-methoxy-6-[3-(trifluoromethyl)phenoxy] picolinic acid (compound No. VI-1)
(1) 2-chloro pyridine N-oxide hydrochloride (17.0 g, 0.102 mol) was mixed with sulfuric acid (64.0 g, 0.102*6.4 mol) and fuming nitric acid (36.0 g (ca. 94%), 0.102*5.3 mol), and the obtained mixture was stirred at a temperature of 90 to 100° C. for 2.5 hours. The obtained reaction mixture was added to 800 ml of ice water to form a precipitate. The precipitate was filtered out, washed with water and then dried. The water phase was extracted with ethyl acetate. The obtained extract was recrystallized with ethyl acetate and hexane. Yield weight: 14.4 g; yield percentage: 81%; solid; melting point: 151 to 153° C.; 1 H-NMR (60 MHz, CDCl3, δ): 7.7-8.2(1H, multi.), 8.2-8.6(2H, complex). |
|
With sulfuric acid; nitric acid In (2S)-N-methyl-1-phenylpropan-2-amine hydrate |
2.1 (1)
(1) Production of 2-chloro-4-nitro pyridine N-oxide as an intermediate 2-chloro pyridine N-oxide hydrochloride (17.0 g, 0.102 mol) was mixed with sulfuric acid (64.0 g, 0.102*6.4 mol) and fuming nitric acid (36.0 g (ca. 94%), 0.102*5.3 mol), and the obtained mixture was stirred at a temperature of 90 to 100° C. for 2.5 hours. The obtained reaction mixture was added to 800 ml of ice water to form a precipitate. The precipitate was filtered out, washed with water and then dried. The water phase was extracted with ethyl acetate. The obtained extract was recrystallized with ethyl acetate and hexane. Yield weight: 14.4 g; yield percentage: 81%; solid; melting point: 151 to 153° C.; 1H-NMR (60 MHz, CDCl3, δ): 7.7-8.2(1H, mult.), 8.2-8.6(2H, complex). |
Reference:
[1]Current Patent Assignee: KUREHA CORP. - US6339045, 2002, B1
Location in patent: Referential example 2
[2]Current Patent Assignee: GLAXOSMITHKLINE PLC - US2002/91264, 2002, A1
[3]Current Patent Assignee: KUREHA CORP. - US6159901, 2000, A
[4]Current Patent Assignee: KUREHA CORP. - US6200933, 2001, B1
- 15
-
[ 14432-16-7 ]
-
[ 929-06-6 ]
-
[ 643744-12-1 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 90℃; for 1h; |
2
2-CHLORO-4-NITROPYRIDINE-1-OXIDE] (Jain, P. [C.] ; Chatterjee, S. K.; Anand, N. Indian Journal of Chemistry 1966, 403) (3.0 g, 17.2 [MMOL),] [2- (2-AMINOETHOXY) ETHANOL] (3.0 g, 28.7 [MMOL)] and [NAHCO3] (2.8 g, 34.4 [MMOL)] were dissolved in ter-amyl alcohol (30 mL) and heated to 90 [XB0;C] for 1 hour. The reaction mixture was then allowed to warm to room temperature, diluted with [CH2CI2] (70 mL) filtered and evaporated to dryness. The crude material thus obtained was purified by flash column chromatography on 230-400 mesh silica gel, utilising as eluent a mixture of EtOAc/MeOH 95: 5 to yield 3.6 g of the title compound. [IR,'H] nmr spectra and mass spectra were consistent with the assigned structure. [CGH13N305 MW =] 243.22 |
Reference:
[1]Current Patent Assignee: GLAXOSMITHKLINE PLC - WO2004/4715, 2004, A2
Location in patent: Page/Page column 19
- 16
-
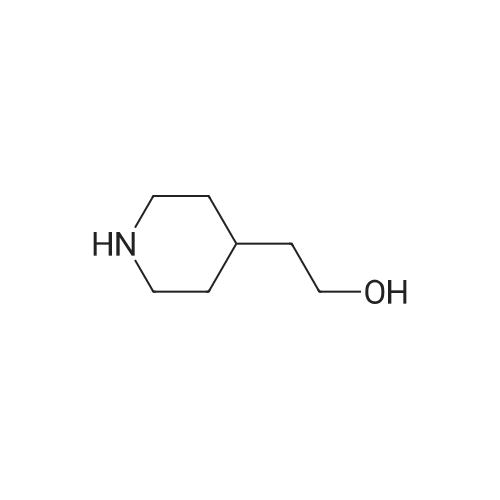 [ 622-26-4 ]
[ 622-26-4 ]
-
[ 14432-16-7 ]
-
[ 643744-13-2 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 90℃; for 1h; |
3
2-CHLORO-4-NITROPYRIDINE-1-OXIDE] (Jain, P. C.; Chatterjee, S. K.; Anand, N. Indian Journal of Chemistry 1966, 403) (2.0 g, 11.5 [MMOL),] 2-piperidin-4-ylethanol (2.0 mL, 15.5 [MMOL)] and [NAHCO3] (1.9 g, 23.0 [MMOL)] were dissolved in ter-amyl alcohol (30 mL) and heated to 90 [XB0;C] for 1 hour. The reaction mixture was then allowed to warm to room temperature and filtered. After dilution with EtOAc (10 mL) and Et2O (3 mL) the desired compound precipitated and was recovered by suction filtration to yield 3.0 g of the title compound as a red solid. [IR,'H] nmr spectra and mass spectra were consistent with the assigned structure. [C12H17N304] [MW =] 267.29 |
Reference:
[1]Current Patent Assignee: GLAXOSMITHKLINE PLC - WO2004/4715, 2004, A2
Location in patent: Page/Page column 19-20
- 17
-
[ 14432-16-7 ]
-
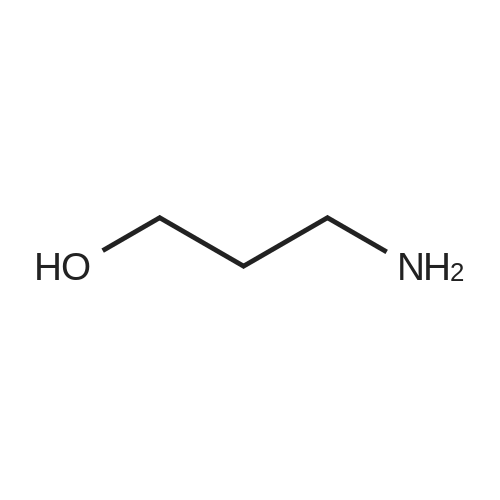 [ 156-87-6 ]
[ 156-87-6 ]
-
[ 205676-72-8 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 90℃; for 0.75h; |
1
2-Chloro-4-nitropyridine-1-oxide (Jain, P. C.; Chatterjee, S. K.; Anand, N. Indian Journal of Chemistry 1966,403) (4.1 g, 23.5 [MMOL),] [3-AMINOPROPAN-1-OL] (3.6 mL, 47.0 [MMOL)] and [NAHCO3] (9.9 [G,] 117.5 [MMOL)] were dissolved in ter-amyl alcohol (35 mL) and heated to 90 [XB0;C] for 45 minutes. The reaction mixture was then allowed to warm to room temperature, diluted with [CH2CI2] (85 mL) filtered and evaporated to dryness. The crude material thus obtained was purified by gradient flash column chromatography on 230- 400 mesh silica gel, utilising a mixture of EtOAc/MeOH 90: 10 as starting eluent, and a mixture of EtOAc/MeOH 70: 30 as final eluent, to yield 4.4 g of the title compound. [IR, 1H] nmr spectra and mass spectra were consistent with the assigned structure. [C8H11N304] MW = 213. 20 |
Reference:
[1]Current Patent Assignee: GLAXOSMITHKLINE PLC - WO2004/4715, 2004, A2
Location in patent: Page/Page column 19
- 18
-
[ 887278-70-8 ]
-
[ 14432-16-7 ]
-
[ 890301-68-5 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With 4-methyl-morpholine In methanol at 80℃; |
12
2-Chloro-4-nitropyridine N-oxide (0.99 g, 5.67 mmol), methyl 4'-(aminomethyl)-3,3'-difluorobiphenyl-2-carboxylate (1.05 g, 3.78 mmol), and N-methylmorpholine (1.15 g, 11.35 mmol) were suspended in methanol (7.6 mL) under nitrogen. The reaction vessel was sealed and placed into a 80° C. oil bath for overnight heating and stirring. After 15 hours, the reaction mixture was cooled and concentrated. The residue was subjected to silica gel chromatography eluted with 50% ethyl acetate in hexane to give methyl 3,3'-difluoro-4'-[(4-nitro-1-oxidopyridin-2-yl)amino]methyl}biphenyl-2-carboxylate. To a solution of the above material (0.88 g, 2.11 mmol) in methanol (35 mL) was added N-methylmorpholine (0.32 g, 3.17 mmol). Methanethiol gas was bubbled through the solution until saturation. The reaction vessel was sealed and placed in a 100° C. oil bath for 12 hours. The reaction mixture was cooled and concentrated. Silica gel chromatography with 1-10% methanol in methylene chloride provided methyl 3,3'-difluoro-4'-([4-(methylthio)-1-oxidopyridin-2-yl]amino}methyl)biphenyl-2-carboxylate. To a solution of the above material (0.72 g, 1.73 mmol) in chloroform (15 mL), at 0° C., was added mCPBA (0.39 g, 2.25 mmol) slowly over 13 minutes. After 50 minutes, calcium hydroxide (0.26 g, 3.46 mmol) was added, and the resulting mixture was stirred for 15 minutes. The mixture was filtered and the filtrate was concentrated to give methyl 3,3'-difluoro-4'-([4-(methylsulfinyl)-1-oxidopyridin-2-yl]amino}methyl)biphenyl-2-carboxylate. To a solution of the above material (0.748 g, 1.73 mmol) in methanol (27 mL) was added a 50% aqueous slurry of Raney Nickel (approximately 1 mL). The reaction vessel was purged with nitrogen and then flushed with hydrogen from a balloon. After 2 hours, the reaction mixture was purged with nitrogen prior to filtration through a pad of celite. The filtrate was concentrated to an oily residue, which was subjected to silica gel chromatography eluted with 1-6% methanol in methylene chloride to provide methyl 3,3'-difluoro-4'-([4-(methylsulfinyl)pyridin-2-yl]amino}methyl)biphenyl-2-carboxylate. A solution of the above material (0.10 g, 0.24 mmol) in trifluoroacetic anhydride (3 mL) was heated at 50° C. for 3 hours. The mixture was concentrated, and the residue was dissolved in a 1:1 mixture (4 mL) of triethylamine and methanol. After 10 minutes, the solution was concentrated. The residue was again dissolved in methanol and concentrated (×4) to afford methyl 3,3'-difluoro-4'-[(4-mercaptopyridin-2-yl)amino]methyl}biphenyl-2-carboxylate as an oil, which was used directly in the next reaction without purification. A solution of the above material (93 mg, 0.24 mmol) in a mixture of ethyl acetate (6 mL) and water (6 mL) was cooled to 0° C. Chlorine gas was bubbled through the solution for 1 minute. The bright yellow solution was partitioned between methylene chloride (80 mL) and water (80 mL), and the organic layer was dried over sodium sulfate, filtered, and concentrated. The oily residue was then dissolved in methylene chloride (4 mL) and cooled to 0° C. 1-(2-Aminoethyl)piperidine (0.19 g, 1.45 mmol) was added, and the resulting mixture was stirred at 0° C. for 1 hour and then warmed to room temperature for overnight stirring. The mixture was subjected to silica gel chromatography eluted with 1-5% methanol in methylene chloride to provide methyl 4'-[(3,5-dichloro-4-[(2-piperidin-1-ylethyl)-amino] sulfonyl}pyridin-2-yl)amino]methyl}-3,3'-difluorobiphenyl-2-carboxylate. A solution of the above material (40 mg, 0.07 mmol) in 2M ammonia in methanol (3 mL) was purged with nitrogen, and 10% Pd/C (16 mg) was added. The reaction vessel was purged with nitrogen and then flushed with hydrogen from a balloon. Additional 10% Pd/C (10 mg) was added every hour for 5 hours until the reaction reached 80% completion. The reaction mixture was purged with nitrogen and then filtered through a pad of celite. The filtrate was concentrated, and the residue was purified by silica gel chromatography eluted with 1-6% methanol in methylene chloride to provide the title compound. HRMS (M+H+): calc'd 545.2029, found 545.2047. 1HNMR (CD3OD, 400 MHz): δ 8.15 (1H, d, J=5.6 Hz), 7.55 (1H, m), 7.43 (1H, t, J=7.6 Hz), 7.26-7.20 (2H, m), 7.12-7.09 (2H, m), 6.96 (1H, bs), 6.88 (1H, bd, J=5.2 Hz), 4.66 (2H, s), 3.67 (3H, s), 3.03 (2H, t, J=7.2 Hz), 2.40 (2H, t, J=7.2 Hz), 2.36 (4H, bs), 1.54 (4H, m), 1.43 (2H, m) |
Reference:
[1]Current Patent Assignee: Wood, Michael R.; Bock, Mark G.; Books, Kathy M.; Freidinger, Roger M.; Kim, June J. - US2006/122236, 2006, A1
Location in patent: Page/Page column 17-18
- 19
-
[ 14432-16-7 ]
-
[ 75-04-7 ]
-
2-ethylamino-4-nitropyridine N-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With triethylamine In ethanol for 4h; Heating / reflux; |
|
|
With triethylamine In ethanol for 4h; Heating / reflux; |
1
2.0 g of 2-chloro-4-nitro-pyridine 1-oxide is dissolved in 20 ml ethanol. 11.5 ml triethylamine is added and it is stirred for 4 hours under reflux. The solution is condensed off on the rotary evaporator. After purification by chromatography on silica gel, 1.5 g of title compound is obtained. 1H-nMR (DMSO-d6): δ=1.19 (t, 3H); 3.39 (pentuplet, 2H); 7.39 (dd, 1H); 7.47 (d, 1H); 7.64 (t, 1H); 8.35 (d, 1H) ppm. |
Reference:
[1]Current Patent Assignee: BAYER AG - WO2006/82107, 2006, A1
Location in patent: Page/Page column 41
[2]Current Patent Assignee: BAYER AG - US2007/15759, 2007, A1
Location in patent: Page/Page column 37
- 20
-
[ 14432-16-7 ]
-
[ 38608-87-6 ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium methylate In methanol; ethyl acetate |
P.10.2 Production of 4-methoxy-6-[3-(trifluoromethyl)phenoxy] picolinic acid (compound No. VI-1)
(2) 2-chloro-4-nitro pyridine N-oxide (13.4 g, 0.077 mol) was suspended in 100 ml of methanol. Sodium methoxide [14.8 g (ca. 28% methanol solution), 0.077*1.0 mol] was dropped into the obtained suspension and dissolved therein at room temperature while stirring, and the suspension was further stirred for 2 days. The obtained reaction solution was distilled under reduced pressure to remove methanol therefrom. The distillation residue was dissolved in ethyl acetate. The obtained solution was filtered to remove sodium nitrite therefrom, and then ethyl acetate was distilled off, thereby obtaining an aimed product. Yield weight: 12.1 g; yield percentage: 99%; solid; decomposition point: about 90° C.; 1 H-NMR (60 MHz, CDCl3, δ): 3.80(3H, s), 6.75(1H, dd, J=3.5 Hz, 7.5 Hz), 6.99(1H, d, J=3.5 Hz), 8.21(1H, d, J=7.5 Hz). |
|
With sodium methylate; sodium In methanol |
2.1 (1)
(1) 2-Chloro-4-methoxypyridine-1-oxide Sodium (0.46 g) is dissolved in absolute methanol (50 ml) and the resultant solution of sodium methoxide is added to a solution of 2-chloro-4-nitropyridine-1-oxide (3.5 g, prepared as described in Example 1) in methanol (10 ml). The reaction mixture is allowed to stand at room temperature for 50 hours and is then subjected to rotary evaporation to give 2-chloro-4-methoxypyridine-1-oxide. |
|
With sodium methylate In methanol; ethyl acetate |
2.2 (2)
(2) Production of 2-chloro-4-methoxy pyridine N-oxide as an intermediate 2-chloro-4-nitro pyridine N-oxide (13.4 g, 0.077 mol) was suspended in 100 ml of methanol. Sodium methoxide [14.8 g (ca. 28% in methanol solution), 0.077*1.0 mol] was dropped into the obtained suspension and dissolved therein at room temperature while stirring, and the suspension was further stirred for 2 days. The obtained reaction solution was distilled under reduced pressure to remove methanol therefrom. The distillation residue was dissolved in ethyl acetate. The obtained solution was filtered to remove sodium nitrite therefrom, and then ethyl acetate was distilled off, thereby obtaining an aimed product. Yield weight: 12.1 g; yield percentage: 99%; solid; decomposition point: about 90° C.; 1H-NMR (60 MHz, CDCl3, δ): 3.80(3H, s), 6.75(1H, d, J=3.5 Hz, 7.5 Hz), 6.99(1H, d, J=3.5 Hz), 8.21(1H, d, J=7.5 Hz). |
Reference:
[1]Current Patent Assignee: KUREHA CORP. - US6159901, 2000, A
[2]Current Patent Assignee: BOSTON SCIENTIFIC CORP - US4587240, 1986, A
[3]Current Patent Assignee: KUREHA CORP. - US6200933, 2001, B1
- 24
-
[ 14432-16-7 ]
-
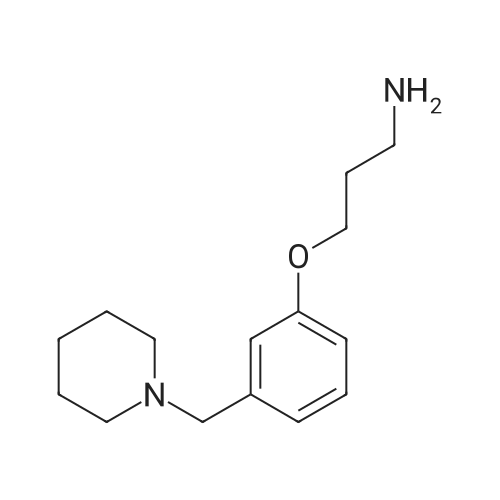 [ 73278-98-5 ]
[ 73278-98-5 ]
-
4-nitro-2-[3-(3-piperidin-1-ylmethylphenoxy)propylamino]-1-oxypyridine
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With triethylamine In ethanol |
6.i 2-[3-[3-(Piperidinomethyl)phenoxy]propylamino]pyrid-4-one
(i) 3-[3-(Piperidinomethyl)phenoxy]propylamine (6.21 g), 2-chloro-4-nitropyridine-N-oxide (4.36 g) and triethylamine (6.32 g) were stirred, in ethanol (30 ml), under reflux for 31/2 hours. The reaction mixture was cooled gradually to 0° C. to yield 2-[3-[3-(piperidinomethyl)phenoxy]propylamino]-4-nitropyridine-N-oxide (4.23 g), m.p. 90°-90.5° C. |
|
With triethylamine In ethanol |
13.i 2-[3-[3-(Piperidinomethyl)phenoxy]propylamino-4-benzyloxypyridine
(i) 3-[3-(Piperidinomethyl)phenoxy]propylamine 6.21 g), 2-chloro-4-nitropyridine-N-oxide (4.36 g) and triethylamine (6.32 g) were stirred, in ethanol (30 ml), under reflux for 31/2 hours. The reaction mixture was cooled gradually to 0° C. to yield 2-[3-[3-(piperidinomethyl)phenoxy]propylamino]-4-nitropyridine-N-oxide (4.23 g), m.p. 90°-90.5° C. |
Reference:
[1]Current Patent Assignee: GLAXOSMITHKLINE PLC - US4608380, 1986, A
[2]Current Patent Assignee: GLAXOSMITHKLINE PLC - US4681883, 1987, A
- 28
-
[ 14432-16-7 ]
-
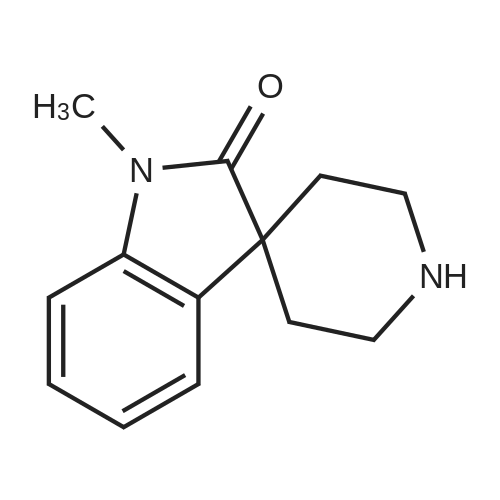 [ 67677-81-0 ]
[ 67677-81-0 ]
-
C18H18N4O4
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With N-ethyl-N,N-diisopropylamine In 1-Methylpyrrolidine at 48℃; for 48h; |
94
Examples 94-97H2N MThe appropriate piperidine (150μmol, 5eq) was dissolved in N-methylpyrrolidine (100μL), N1N- diisopropylethylamine (150μmol, 5eq) was added followed by a solution of 2-chloro-4- nitropyridine N-oxide (30μmol, 1eq) in N-methylpyrrolidine (200μL). The reaction mixture was sealed and heated at 480C for 48 hours. The reaction mixture was concentrated in vacuo then redissolved in methanol (100μL). A solution 1 M solution of ammonium formate in methanol (400μL) was added followed by 10% palladium on carbon (approx 10% by weight). The reaction mixture was sealed and shaken at room temperature overnight. The reaction mixture was decanted, discarding the catalyst and the methanol evaporated. The material was purified by HPLC on a Phenomenex Luna C18, 10μm, 150 x 10 mm id column using acetonitrile: 0.05% aqueous diethylamine as the mobile phase. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 103-104
- 29
-
[ 14432-16-7 ]
-
[ 909035-25-2 ]
-
[ 937255-01-1 ]
| Yield | Reaction Conditions | Operation in experiment |
| 42% |
With pyridine; N-ethyl-N,N-diisopropylamine In tert-Amyl alcohol for 1h; Heating / reflux; |
116
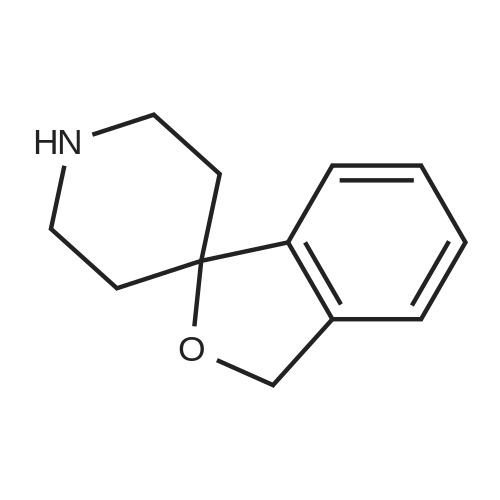
Preparation 1166-Fluoro-1'-(4-nitro-1-oxidopyridin-2-yl)spirori-benzofuran-3,4'-piperidinelThe piperidine of Preparation 66 (167mg, O.δmmol), 2-chloro-4-nitropyridine N-oxide (150mg, 0.89mmol), N,N-diisopropylethylamine (150μL, 0.86mmol), and pyridine (5mg) were combined in 2-methyl-2-butanol (8ml) and heated at reflux for 1 hour. The reaction mixture was diluted with pentane (25ml) and filtered through a silica plug. The title compound was eluted with ether. The material was collected as an orange solid (117mg, 0.34mmol, 42%). 1H-NMR (CD3OD, 400MHz): δ 1.85 (m, 2H), 2.2 (m, 2H), 3.0 (t, 2H), 3.95 (m, 2H), 4.6 (s, 2H), 6.5 (d, 1H), 6.6 (m, 1 H), 7.2 (m, 1H), 7.8 (m, 1H), 7.9 (s, 1 H), 8.4 (d, 1 H). LRMS m/z (APCI) 346 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 178
- 30
-
[ 14432-16-7 ]
-
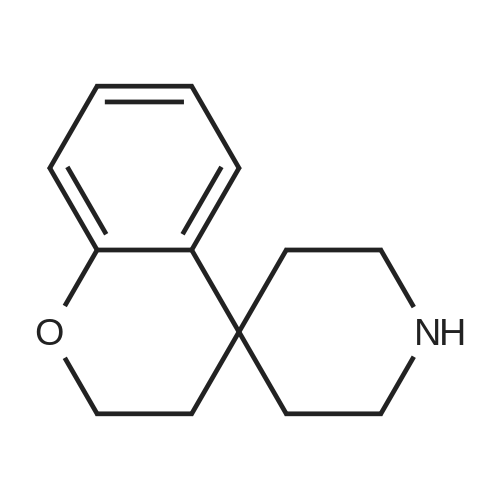 [ 147373-05-5 ]
[ 147373-05-5 ]
-
C18H19N3O4
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 48℃; for 16h; |
120
Example 1202-(2.3-Dihvdro-1η-spirofchromene-4λ'-piperidin]-1 '-yl)pyridin-4-amine2,3-Dihydrospiro[chromene-4,4'-piperidine] [described in J. Med. Chem. 1995, 38, (11), 2009- 2017] (0.8g, 1.36mmol), 2-chloro-4-nitropyridine N-oxide (226mg, 1.29mmol) and sodium hydrogen carbonate (171 mg, 2.0mmol) were combined in 2-methyl-2-butanol (4ml). The reaction mixture was heated at 480C for 16 hours. The reaction mixture was concentrated in vacuo. The residue was extracted from water into dichloromethane. The crude material was redissolved in acetic acid (5ml). Iron powder (143mg, 2.55mmol) was added and the reaction mixture stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue extracted from 2M sodium hydroxide into dichloromethane. The combined organic extracts were dried over sodium sulphate, filtered and evaporated to yield the crude aminopyridine. The material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: 0.88 ammonia (95:5:0.5) to yield the title compound (155mg, 0.52mmol).1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1 H), 1.7 (m, 2H), 2.1 (m, 2H), 2.2 (m, 2H), 3.0 (m, 2H), 4.0 (m, 2H), 4.2 (m, 3H), 5.9 (s, 1 H), 6.0 (d, 1 H), 6.8-7.2 (m, 3H), 7.3 (m, 1 H), 7.9 (m, 1 H). LRMS m/z (APCI) 296 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 111
- 31
-
[ 14432-16-7 ]
-
spiro[1-benzofuran-3,4'-piperidine]-7-carbonitrile hydrochloride
[ No CAS ]
-
[ 937255-08-8 ]
| Yield | Reaction Conditions | Operation in experiment |
| 58% |
With sodium hydrogencarbonate In tert-Amyl alcohol at 50℃; for 48h; |
123
Preparation 1231 '-(4-nitro-1 -oxidopyridin-2-yl)spirori -benzofuran-3,4'-piperidinel-7-carbonitrileThe piperidine of Preparation 55 (565 mg, 2.28 mmol) was dissolved in 2-methyl-2-butanol (10ml). 2-chloro-4-nitropyridine N-oxide (398 mg, 2.28 mmol) and sodium hydrogen carbonate (300 mg 3.57 mmol) were added and the reaction was heated at 5O0C for 48 hours. The reaction mixture was concentrated in vacuo and the residue taken up in ethyl acetate (15ml). The organic phase was washed with water (3 x 10ml) and then dried over sodium sulfate. The material was purified by column chromatography using an ISCO silica cartridge eluting with a gradient of heptane to 70:30 heptane/ethyl acetate to yield the title compound as a yellow solid (459 mg, 1.30 mmol, 58%).1H-NMR (CDCI3, 400MHz): δ 1.9-1.95 (d, 2H), 2.10-2.15 (m, 2H), 3.0-3.05 (m, 2H), 3.95-4.05 (m, 2H), 5.75 (s, 2H), 7.05 (t, 1 H), 7.40 (d, 1 H), 7.55 (d, 1 H), 7.8 (m, 1 H), 7.90 (m, 1 H), 8.4 (d, 1H).LRMS m/z (APCI) 353 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 182
- 32
-
[ 14432-16-7 ]
-
[ 937255-03-3 ]
-
C23H28N4O4
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 48℃; for 18h; |
155
Example 155(3S)-1'-(4-aminopyridin-2-yl)-N,N-diethyl-2,3-dihvdrospiro[indene-1 ,4'-piperidinel-3- carboxamideThe piperidine of Preparation 118 (92mg. 0.3mmol), 2-chloro-4-nitropyridine N-oxide (50mg, 0.28mmol) and sodium hydrogen carbonate (28mg, 0.33mmol) were combined in 2-methyl-2- butanol (2ml) and the reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (113mg, 1.8mmol) and palladium hydroxide (11 mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/ methanol/0.880 ammonia (95:5:0.5) as eluent to afford the title compound as a gum (67mg, 0.18mmol, 56%).1H-NMR (CDCI3, 400MHz): δ 1.2 (m, 3H), 1.3 (m, 3H), 1.6 (m, 1H), 1.8 (m, 2H), 2.2 (m, 1 H)1 2.4 (m, 1 H), 2.5 (m, 1 H), 3.0 (m, 2H), 3.4 (m, 1 H), 3.5 (m, 1 H), 3.6 (m, 2H), 3.9 (s, 2H), 4.1 (m, 1H), 4.3 (m, 2H), 5.9 (s, 1H), 6.0 (d, 1H), 7.1 (m, 1H), 7.2 (m, 3H), 7.9 (d, 1H). LRMS m/z (ESI) 379 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 121
- 33
-
[ 14432-16-7 ]
-
[ 937255-05-5 ]
-
C21H23N3O7
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In isopropyl alcohol at 47℃; for 20h; |
156.a
Example 156(a) Ethyl r-(4-aminopyridin-2-yl)-5-methoxy-3H-spiro[2-benzofuran-1 ,4'-piperidine1-3- carboxylate The amine of Preparation 120 (1.57g, 5.39 mmol), 2-chloro-4-nitropyridine N-oxide (941 mg, 5.39 mmol) and NaHCO3 (498mg, 5.93 mmol) were stirred in isopropyl alcohol at 470C under nitrogen for 20 hours. The solvent was evaporated, giving the crude 2-amino-4-nitropyridine- N-oxide intermediate as a yellow gum. This gum was partially purified by flash column chromatography eluting with ethyl acetate/heptane (1:1) followed by dichloromethane/methanol (19:1), collecting and evaporating the yellow coloured fractions which on evaporation gave a yellow foam. This foam was treated with ammonium formate (2.2g) and Pd(OH)2 (223mg) and heated in ethanol (20ml) at 7O0C for 1 hour. Further additions of ammonium formate (2.2g) and Pd(OH)2 (223mg) were made, and heating continued for 1 further hour. The solution was filtered through Arbocel and evaporated. This crude material was purified by flash column chromatography eluting with dichloromethane/methanol/0.880 ammonia (96.7:3:0.3) to give the title compound as a white foam (1.3g, 63%).1H-NMR (CDCI3, 400MHz): δ 1.3 (t, 3H), 1.80 (m, 1 H), 1.95 (m, 3H), 3.4 (m, 2H), 3.8 (s, 3H), 4.0 (S, 2H), 4.2 (m, 4H), 5.65 (s, 1H), 5.95 (d, 1H), 6.0 (m, 1 H), 6.85 (m, 1 H), 6.95 (d, 1 H), 7.0 (m, 1 H), 7.9 (d, 1 H). LRMS m/z (ES) 384 [MH]+ |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 121-122
- 34
-
[ 14432-16-7 ]
-
[ 937255-12-4 ]
-
[ 937255-15-7 ]
| Yield | Reaction Conditions | Operation in experiment |
| 79% |
With triethylamine In tert-Amyl alcohol at 50℃; |
130
Preparation 130Methyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H)-carboxylateThe compound of Preparation 127 (340 mg, 1.38 mmol) was stirred with 2-chloro-4- nitropyridine-N-oxide (361 mg, 2.07 mmol) and triethylamine (0.29 ml, 2.07 mmol) at 500C in t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles removed in vacuo. The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow foam (420 mg, 1.09 mmol, 79%).1H-NMR (CDCI3, 400MHz): δ 1.65 (m, 2H), 2.24 (m, 2H), 2.96 (m, 2H), 3.85-4.06 (m, 6H), 7.03 (t, 1 H), 7.20 (d, 1 H), 7.25 (t, 1 H), 7.68-7.74 (m, 2H), 7.90 (m, 1 H), 8.26 (d, 1 H). MS m/z (APCI) 385 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 185-186
- 35
-
[ 14432-16-7 ]
-
[ 937255-13-5 ]
-
[ 937255-16-8 ]
| Yield | Reaction Conditions | Operation in experiment |
| 87% |
With triethylamine In tert-Amyl alcohol at 50℃; |
131
Preparation 131Ethyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H)-carboxylateThe compound of Preparation 128 (350 mg, 1.34 mmol) was stirred with 2-chloro-4- nitropyridine-N-oxide (352 mg, 2.02 mmol) and triethylamine (0.28 ml, 2.02 mmol) at 500C in t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles removed in vacuo.The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow foam (465 mg, 1.17 mmol, 87%).1H-NMR (CDCI3, 400MHz): δ 1.39 (m, 3H), 1.65 (m, 2H), 2.25 (m, 2H), 2.97 (m, 2H), 3.94-4.06 (m, 4H), 3.32 (m, 2H), 7.03 (t, 1 H), 7.20 (d, 1H), 7.24 (t, 1 H), 7.68-7.74 (m, 2H), 8.25 (m,1 H). MS m/z (APCI) 399 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 186
- 36
-
[ 14432-16-7 ]
-
[ 937255-14-6 ]
-
[ 937255-17-9 ]
| Yield | Reaction Conditions | Operation in experiment |
| 75% |
With triethylamine In tert-Amyl alcohol at 50℃; |
132
Preparation 132 Isopropyl 1 '-(4-nitro-1 -oxidopyridin-2-v0spirorindole-3,4'-piperidinel-1 (2H VcarboxylateThe compound of Preparation 129 (230 mg, 0.84 mml) was stirred with 2-chloro-4- nitropyridine-N-oxide (219 mg, 1.26 mmol) and triethylamine (0.18 ml, 1.26 mmol) at 500C in t-amyl alcohol (5 ml) overnight. The mixture was cooled and the volatiles were removed in vacuo. The residue was resuspended in toluene, dried (MgSO4) and filtered. The filtrate was loaded onto a silica gel column and eluted with cyclohexane:ethyl acetate (100:0 - 0:100) to yield the title compound as a yellow glass (260 mg, 0.63 mmol, 75%).1H-NMR (CDCI3, 400MHz): δ 1.39 (m, 6H), 1.65 (m, 2H), 2.23 (m, 2H), 2.97 (m, 2H), 3.93- 4.07 (m, 4H), 5.12 (m, 1 H), 7.02 (t, 1 H), 7.20 (d, 1 H), 7.23 (t, 1 H), 7.71 (d, 1 H), 7.74 (m, 1 H), 7.90 (m, 1 H), 8.26 (d, 1 H). MS m/z (APCI) 413 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 187
- 39
-
[ 14432-16-7 ]
-
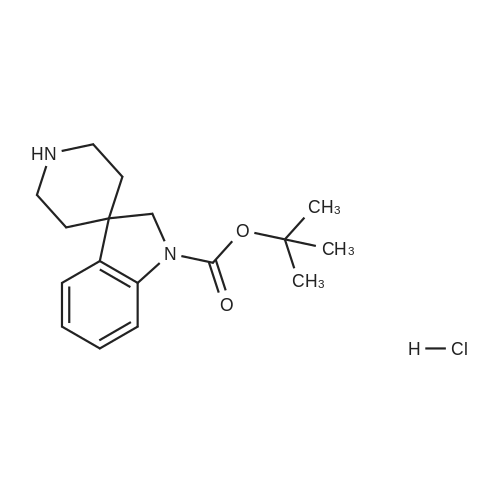 [ 1243474-66-9 ]
[ 1243474-66-9 ]
-
C22H26N4O5
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate; triethylamine; In tert-Amyl alcohol; at 48.0℃; for 18.0h; |
Example 154 2-M .2-Dihvdro-i 'H-spiro?ndole-3,4'-piperidin1-1 '-yl)pyridin-4-amine(a) Tert-butyl r-(4-aminopyridin-2-yl)spirorindole-3,4'-piperidine1-1 (2H)-carboxylateTert-butyl spiro[indole-3,4'-piperidine]-1(2H)-carboxylate. HCI (1g, 3.1 mmol) [described in WO2004/028459, example 21 , compound xxii], 2-chloro-4-nitropyridine N-oxide (538mg, 2.94mmol), sodium hydrogen carbonate (310mg, 3.72mmol) and triethylamine (0.472ml, 3.4mmol) were combined in 2-methyl-2-butanol (15ml) and the reaction mixture heated at 480C for 18 hours. After this time ammonium formate (1.16g, 18.6mmol) and palladium hydroxide (100mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane: methanol: 0.880 ammonia (90:10:1) as eluent to afford the title compound as a beige foam (1.1g, 2.89mmol,1H-NMR (CDCI3, 400MHz): delta 1.6 (s, 10H), 1.8 (m, 2H), 2.0 (m, 2H), 2.9 (m, 2H), 3.9 (bs, 2H), 4.1 (bs, 1 H), 4.2 (m, 2H), 5.9 (s, 1 H), 6.05 (d, 1 H), 7.0 (t, 1 H), 7.1 (d, 1 H), 7.2 (t, 1 H), 7.9 (d, 1 H). LRMS m/z (APCI) 381 [MH]+. |
Reference:
[1]Patent: WO2007/57775,2007,A1 .Location in patent: Page/Page column 120
- 40
-
[ 14432-16-7 ]
-
[ 937254-20-1 ]
-
C19H19N3O6
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 48℃; for 72h; |
39.a
Example 39(a) Methyl 1 '-(4-aminopyridin-2-yl)spirori-benzofuran-3.4'-piperidine1-2-carboxylateThe piperidine of Preparation 19 (569mg,,2.3mmol), 2-chloro-4-nitropyridine N-oxide (384mg, 2.2mmol) and sodium hydrogen carbonate (285mg, 3.44mmol) were combined in 2-methyl-2- butanol (10ml) and the reaction mixture was heated at 480C for 72 hours. After this time ammonium formate (1.48g, 23.6mmol) and palladium hydroxide (152mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen. The reaction mixture was filtered through Arbocel and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/ methanol/0.880 ammonia (90:10:1) as eluent to afford the title compound as a brown solid (560mg, 1.65mmol, 72%).1H-NMR (CDCI3, 400MHz): δ 1.8 (m, 1 H), 2.0 (m, 3H), 3.5 (m, 2H), 3.6 (m, 2H), 3.8 (m, 1 H), 3.8 (s, 3H), 4.0 (m, 4H), 5.0 (s, 1 H), 5.9 (s, 1H), 6.0 (d, 1 H), 6.9 (m, 2H), 7.2 (m, 2H), 7.9 (d, 1 H). LRMS m/z (APCI) 340 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 97
- 41
-
[ 14432-16-7 ]
-
[ 937254-22-3 ]
-
C25H23ClN4O5
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 50℃; |
16
Example 16(2f?)-r-(4-Aminopyridiϖ-2-yl)-N-(3-chlorobenzvϖspiroϖ-benzofuran-3,4'-piperidinel-2- carboxamideThe piperidine of Preparation 21 (85mg, 0.237mmol), 2-chloro-4-nitropyridine N-oxide (42mg, 0.241 mmol) and sodium hydrogen carbonate (37mg, 0.443mmol) were combined in 2-methyl- 2-butanol (4ml). The reaction mixture was heated at 500C overnight. The solvent was evaporated in vacuo and the residue redissolved in acetic acid (4ml), iron powder (122mg, 2.1 mmol) was added and the reaction mixture stirred at room temperature for 1 hour. The reaction mixture was extracted from 2M sodium hydroxide into ethyl acetate. The combined organic extracts were dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO silica cartridge eluting with dichloromethane/ methanol/0.880 ammonia (95:5:0.5). The product was obtained as a white solid (42mg, 0.09mmol, 39%).1H-NMR (CD3OD, 400MHz): δ 1.8 (m, 1 H), 1.9 (m, 1 H), 2.0 (m, 1 H), 2.1 (m, 1 H), 2.5 (m, 2H), 3.7 (m, 2H), 4.4 (d, 2H), 4.85 (s, 1 H), 6.0 (s, 1 H), 6.1 (d, 1 H), 6.9 (t, 2H), 7.2-7.4 (m, 6H), 7.6 (d, 1 H). LRMS m/z (APCI) 449 [MH]+. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 85
- 43
-
[ 14432-16-7 ]
-
[ 937254-46-1 ]
-
C20H21N3O5
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With sodium hydrogencarbonate In tert-Amyl alcohol at 48℃; for 18h; |
109.a
Example 109 I'-^-Aminopyridin^-vD^^-dihvdrospiroϖndene-I λ'-piperidinel-S-carboxylic acid(a) Methyl (3S)- 1 '-(4-aminopyridin-2-yl')-2.3-dihvdrospirofindene-1 ,4'-piperidinel-3-carboxylateThe piperidine of Preparation 47 (100mg, 0.41 mmol), 2-chloro-4-nitropyridirie N-oxide (68mg, 0.389mmo.) and sodium hydrogen carbonate (68mg, 0.82mmol) were combined in 2-methyl- 2-butanol (2.5ml) and the reaction mixture was heated at 480C for 18 hours. After this time ammonium formate (154mg, 2.46mmol) and palladium hydroxide (10mg) were added and the reaction mixture heated for a further 2 hours at 650C. This was repeated until complete reduction was seen by thin layer chromatography. The reaction mixture was filtered through Arbocel and the combined filtrates concentrated in vacuo. The crude product was purified by column chromatography on silica gel using dichloromethane/methanol/0.880 ammonia (95:5:0.5) as eluent to afford the title compound as a white foam (90mg, 0.26mmol, 63%).1H-NMR (CDCI3, 400MHz): δ 1.6 (m, 1H), 1.8 (m, 1 H), 1.9 (m, 1 H), 2.0 (m, 1 H), 2.4 (d, 2H), 3.0 (m, 2H), 3.8 (s, 3H), 4.0 (bs, 2H), 4.2 (m, 2H), 4.3 (m, 1 H), 5.9 (s, 1 H), 6.0 (d, 1H), 7.2 (m, 3H), 7.4 (d, 1 H), 7.9 (d, 1 H). Rf: 0.35 dichloromethane/methanol/0.880 ammonia (94.5: 5: 0.5). |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 108
- 47
-
[ 14432-16-7 ]
-
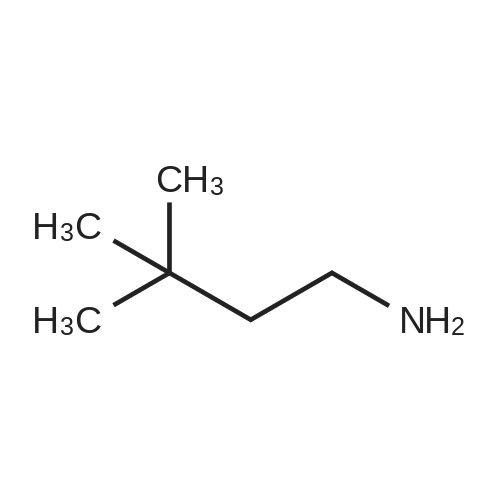 [ 15673-00-4 ]
[ 15673-00-4 ]
-
[ 1041054-28-7 ]
| Yield | Reaction Conditions | Operation in experiment |
|
In ethanol at 80℃; |
VI
2-Chloro-4-nitropyridine 1 - oxidc was dissolved in EtOH and treated with the amine component (2.2 eq) at 800C overnight. After evaporation of the EtOH, the residue was dissolved in DCM and purified. Purification of the intermediate 2-(3,3-dimethylbutylamino)-4-mtropyridine 1 -oxide was carried out using silica gel (gradient: 0 to 100% (EtOAc/DCM 1 : 1 ) in DCM). (Calculated mass: 241 .2, observed mass: 281.0 (M+AcN)+). 1H-NMR (500 MHz, CDCl3) δ (ppm) 8.30 (d, I H), 7.42 (d, I H), 7.40 (s, I H), 6.92 (bs, IH), 3.38 (m, 2H), 1 .66 (t, 2H), 1.02 (s, 9H). Subsequent reduction with Raney nickel and 1 atm hydrogen in MeOH, at r.t. for 3 hr provided the title compound which was used in the next step without further purification. |
Reference:
[1]Current Patent Assignee: KEMIA, INC - WO2008/89034, 2008, A2
Location in patent: Page/Page column 126
- 50
-
[ 14432-16-7 ]
-
[ 937254-70-1 ]
-
[ 937254-71-2 ]
| Yield | Reaction Conditions | Operation in experiment |
| 70% |
Stage #1: 2,3-dihydrospiro[indene-1,4-piperidine]-3-amine With sodium carbonate In 4-methyl-2-pentanone at 110℃; for 3h;
Stage #2: 2-chloro-4-nitropyridine-N-oxide In 4-methyl-2-pentanone at 80℃; |
82
Preparation 82 1'-(4-Nitro-1-oxidopyridin-2-ylV2,3-dihvdrospirorindene-1 ,4'-piperidin1-3-amineThe amine of Preparation 81 (260mg, 0.95mmol) and sodium carbonate (352mg, 3.32mmol) were combined in methyl isobutyl ketone (10ml) and heated at 11O0C for 3 hours. The reaction mixture was cooled to 8O0C before the addition of 2-chIoro-4-nitropyridine N-oxide. The reaction mixture was heated at 8O0C overnight. The reaction mixture was washed with water and the solvent evaporated in vacuo. The residue was redissolved in water / 2-propanol and stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo then partitioned between dichloromethane and water. The organic phase was washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude product was purified by column chromatography using an ISCO silica cartridge eluting with dichloromethane - 90:10:1 dichloromethane/methanol/0.880 ammonia. The product was isolated as an orange gum (230mg, 0.67mmol, 70%).1H-NMR (CD3OD, 400MHz): δ 1.6 (m, 3H), 2.0 (t, 1 H), 2.4 (t, 1 H), 2.8 (m, 1 H), 3.1 (m, 2H), 4.0 (m, 2H), 4.4 (m, 1 H), 7.2 (m, 3H), 7.4 (s, 1 H), 7.8 (s, 1 H), 7.9 (s, 1 H), 8.4 (s, 1 H). |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2007/57775, 2007, A1
Location in patent: Page/Page column 165
- 51
-
[ 23056-36-2 ]
-
[ 14432-16-7 ]
| Yield | Reaction Conditions | Operation in experiment |
| 90% |
Stage #1: 2-chloro-4-nitropyridine With urea hydrogen peroxide adduct; trifluoroacetic acid In dichloromethane at 0 - 20℃; for 72h;
Stage #2: With sodium dithionite In dichloromethane; water for 0.25h;
Stage #3: With hydrogenchloride In dichloromethane; water |
32.A
[241] Step A: 2-Chloro-4-nitropyridine 1 -oxide: Into a mixture of 2-chloro-5-methylpyridine, urea hydrogen peroxide addition compound and dichloromethane was added anhydrous trifluoroacetic acid dropwise at O0C, and the mixture was stirred at O0C for 1 hour. After stirring for 3 days while elevating the reaction temperature to room temperature, an aqueous solution (250 mL) of sodium hydrosulfite (45 g) was added, and the reaction mixture was stirred for 15 minutes. Hydrochloric acid (0.5N, 400 mL) was added and the mixture was extracted with dichloromethane (400 mL). The organic phase was washed with sodium bicarbonate solution, dried over magnesium sulfate, filtered and the solvent removed under reduced pressure to afford the title compound which was used in the next step without further purification as a yellow solid. Yield=90%. 1H NMR (400 MHz, CDCl3): δ= 8.422-8.404 (t, 1 H, J= 3.6 Hz), 8.399-8.375 (t, 1 H, J= 4.8 Hz), 8.067-8.049 (t, 1 H, J= 3.6 Hz). |
| 84% |
With urea hydrogen peroxide adduct; trifluoroacetic anhydride In dichloromethane at 0 - 20℃; for 4.5h; |
2-chloro-4-nitropyridine 1 -oxide (i49):
2-chloro-4-nitropyridine 1 -oxide (i49): To a stirred solution of 2-chloro-4-nitropyridine (5 g, 31.54 mmol) and urea.H202 (6.23 g, 66.20 mmol) in DCM (75 mL), TFAA (13.24 g, 63 mmol) was slowly added at 0°C and the reaction was stirred for 30 min at same temperature thenat room temperature for 4h. Ammonia gas was bubbled into the reaction. The progress of the reaction was monitored by TLC. After completion, the mixture was concentrated under reduced pressure. The crude product was purified by silica gel (100:200 mesh) column chromatography using 1 % methanol in dichloromethane as eluent to afford 2-chloro-4-nitropyridine 1 -oxide (i49) (4.6 g, Yield 84%). 1H NMR (400 MHz, DMSO-d6): δ 8.19 (dd, J = 7.2, 3.2 Hz, 1 H), 8.64 (d, J = 7.2 Hz, 1 H), 8.72 (d, J = 3.2 Hz, 1 H). |
Reference:
[1]Current Patent Assignee: CHEMIZON BEIJING; KAIMEILONG BEIJING PHARMACEUTICAL TECHNOLOGY - WO2010/145197, 2010, A1
Location in patent: Page/Page column 79-80
[2]Current Patent Assignee: UCB - WO2016/124508, 2016, A1
Location in patent: Page/Page column 66
- 52
-
[ 14432-16-7 ]
-
 [ 67385-09-5 ]
[ 67385-09-5 ]
-
C12H17N3O5S
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Stage #1: 2-[(tert-butoxycarbonyl)amino]-1-ethanethiol With sodium hydride In tetrahydrofuran for 0.75h; Inert atmosphere;
Stage #2: 2-chloro-4-nitropyridine-N-oxide In tetrahydrofuran at -78 - 20℃; for 4.75h; Inert atmosphere; |
3.5. Preparation of 2-(4'-nitropyridin-2'-ylsulfanyl)-ethanamine (6k) (4-NO2-2-PSEA)
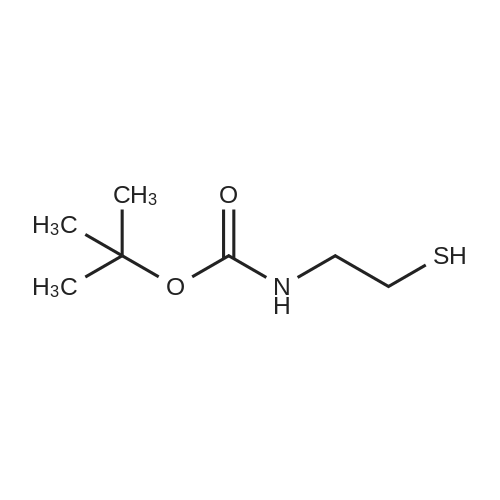
tert-Butyl (2-sulfanylethyl)carbamate (1.38 g, 7.81 mmol) was dissolved in dry THF (20 mL) under a nitrogen atmosphere. NaH (dry, 95%) (0.24 g, 10.2 mmol) was added portion-wise over 45 min and the mixture was then cooled to -78 °C. A solution of 2-chloro-4-nitropyridine N-oxide (1.50 g, 8.59 mmol) in dry THF (40 mL) was added dropwise over 45 min to the cold carbamate solution. The mixture was warmed to ambient temperature over 2 h, stirred for a further 2 h and the THF was removed under reduced pressure. The residue was treated with CHCl3 (50 mL) followed by dropwise addition of PCl3 (3 mL). The reaction mixture was stirred at ambient temperature for 16 h and then poured slowly into water (50 mL). The organic layer was separated and the aqueous layer washed with CHCl3 (4×15 mL). The aqueous layer was basified (pH 8.5-10) with 2 M NaOH solution and extracted several times with CH2Cl2. The combined organic extract was dried (MgSO4), filtered and solvent removed in vacuo to give the title compound 6k as a red-brown oil (ca. 95% pure) (1.06 g, ca. 68%). Preparative TLC (SiO2, CH2Cl2/MeOH/NH3 in MeOH, 9:0.8:0.2) using a sample of crude oil gave the pure title compound 6k as a yellow oil. |
Reference:
[1]Location in patent: experimental part
Mountford, Simon J.; Campi, Eva M.; Robinson, Andrea J.; Hearn, Milton T.W.
[Tetrahedron, 2011, vol. 67, # 2, p. 471 - 485]
- 53
-
[ 14432-16-7 ]
-
 [ 2393-23-9 ]
[ 2393-23-9 ]
-
[ 942076-73-5 ]
| Yield | Reaction Conditions | Operation in experiment |
| 61.2% |
In ethanol for 5h; Reflux; |
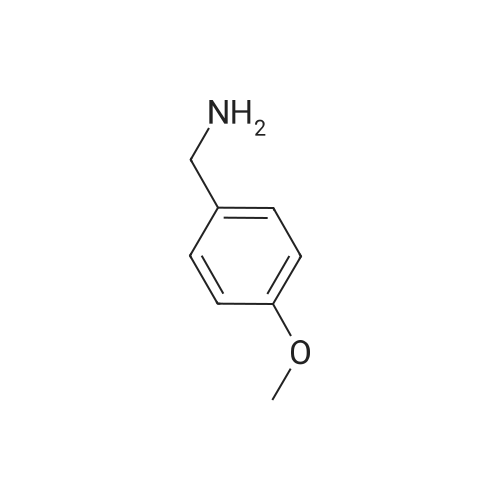
1.1 Step 1
[0060] Step 1: 2-chloro-4-nitro-1-oxo-pyridin-1-ium (40.0 g, 229.2 mmol) and (methoxyphenyl) methanamine (63 g, 458.4 mmol) were dissolved in EtOH (400 mL). The resulting solution was stirred and refluxed for reacting for 5 hours. TLC (PE:EA=2:1) showed that the reaction was completed. Half of the volume of EtOH was concentrated and cooled in an ice bath for 2~3 hours. The cold mixture was filtered and the separated solid was washed with PE (60 mL33) and ice water (60 mL33) respectively, and then dried in vacuum to give N-[(4-(methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin1-ium-2-amine (38.6 g, 140.2 mmol, 61.2% yield) as an orange solid. MS (ESI) Calcd. for C13H13N3O4 275, Found 276 [M+H]+. |
| 40% |
In ethanol for 5h; Reflux; |
|
| 40% |
In ethanol for 5h; Reflux; |
5.16.1
Example 16: 6-({2-[(cyclopropylcarbonyl)amino]pyridin-4-yl}oxy)-iV-hydroxyquinoline-3- carboxamide Compound 1-31 Step 1: 2-(4-methoxybenzyIamino)-4-nitropyridine 1-oxide Iiitermediate-20 [00192] A mixture of 2-chloro-4-nitropyridine- 1 -oxide (75 g, 0.43 mol) and l-(4-methoxyphenyl)- methanamine (125 g, 0.91 mol) in ethanol (1 L) was heated at reflux for 5 h. The reaction was allowed to cool to room temperature and cooled in a freezer overnight. The resulting cold mixture was filtered. The isolated solid was further slurried in methanol (100 mL) and filtered to give N-(4-methoxybenzyl)-4- nitropyridin-2-amine 1-oxide (51.62 g, 40 % yield) as an orange solid. LCMS: (FA) ES+ 276; lH NMR (400 MHz, i¾-DMSO) 5 ppm 8.34 (dd, J= 6.5, 0.8 Hz, 1 H), 8.27 (t, J= 6.7, 6.7 Hz, 1 H), 7.39-7.34 (m, 1 H), 7.29 (d, J= 8.6 Hz, 1 H), 6.89 (d, J= 8.6 Hz, 1 H), 4.52 (d, J= 6.6 Hz, 1 H), 3.70 (s, 1 H). |
| 40% |
In ethanol for 5h; Reflux; |
9.1
Step 1: N-(4-methoxybenzyl)-4-nitropyridin-2-amine 1-oxide Intermediate 19 A mixture of 2-chloro-4-nitropyridine-1-oxide (75 g, 0.43 mol) and 1-(4-methoxyphenyl)-methanamine (125 g, 0.91 mol) in ethanol (1 L) was heated at reflux for 5 h. The reaction was allowed to cool to room temperature and cooled in a freezer overnight. The resulting cold mixture was filtered. The isolated solid was further slurried in methanol (100 mL) and filtered to provide N-(4-methoxybenzyl)-4-nitropyridin-2-amine 1-oxide (51.62 g, 40% yield) as an orange solid. LCMS: (FA) ES+ 276; 1H NMR (400 MHz, d6-DMSO) δ ppm 8.34 (dd, J=6.5, 0.8 Hz, 1H), 8.27 (dd, J=6.7, 6.7 Hz, 1H), 7.39-7.34 (m, 1H), 7.29 (d, J=8.6 Hz, 1H), 6.89 (d, J=8.6 Hz, 1H), 4.52 (d, J=6.6 Hz, 1H), 3.70 (s, 1H). |
Reference:
[1]Current Patent Assignee: WUXI FORTUNE PHARMACEUTICAL - EP3305788, 2018, A1
Location in patent: Paragraph 0059; 0060
[2]Gould, Alexandra E.; Adams, Ruth; Adhikari, Sharmila; Aertgeerts, Kathleen; Afroze, Roushan; Blackburn, Christopher; Calderwood, Emily F.; Chau, Ryan; Chouitar, Jouhara; Duffey, Matthew O.; England, Dylan B.; Farrer, Cheryl; Forsyth, Nancy; Garcia, Khristofer; Gaulin, Jeffery; Greenspan, Paul D.; Guo, Ribo; Harrison, Sean J.; Huang, Shih-Chung; Iartchouk, Natalia; Janowik, Dave; Kim, Mi-Sook; Kulkarni, Bheemashankar; Langston, Steven P.; Liu, Jane X.; Ma, Li-Ting; Menon, Saurabh; Mizutani, Hirotake; Paske, Erin; Renou, Christelle C.; Rezaei, Mansoureh; Rowland, R. Scott; Sintchak, Michael D.; Smith, Michael D.; Stroud, Stephen G.; Tregay, Ming; Tian, Yuan; Veiby, Ole P.; Vos, Tricia J.; Vyskocil, Stepan; Williams, Juliet; Xu, Tianlin; Yang, Johnny J.; Yano, Jason; Zeng, Hongbo; Zhang, Dong Mei; Zhang, Qin; Galvin, Katherine M.
[Journal of Medicinal Chemistry, 2011, vol. 54, # 6, p. 1836 - 1846]
[3]Current Patent Assignee: TAKEDA PHARMACEUTICAL COMPANY LIMITED - WO2011/146591, 2011, A1
Location in patent: Page/Page column 71-72
[4]Current Patent Assignee: TAKEDA PHARMACEUTICAL COMPANY LIMITED - US2012/15942, 2012, A1
Location in patent: Page/Page column 51
- 54
-
[ 14432-16-7 ]
-
 [ 1265355-07-4 ]
[ 1265355-07-4 ]
-
[ 1265361-24-7 ]
| Yield | Reaction Conditions | Operation in experiment |
| 29% |
With caesium carbonate In N,N-dimethyl-formamide at 20℃; for 1h; |
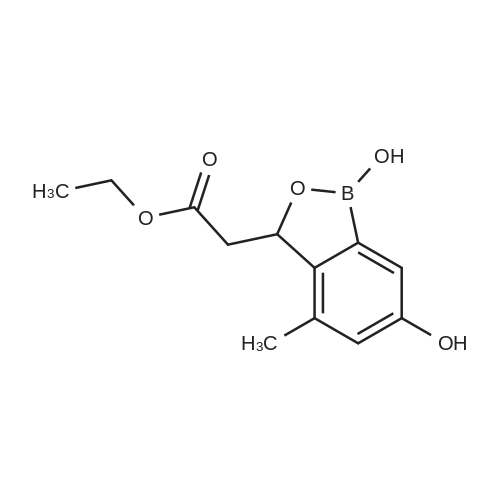
1.G53.1
benzo[c] [ 1 ,2] oxaborol-3-yl] -acetic acid ethyl ester[0517] (l,6-dihydroxy-4-methyl-l,3-dihydro-benzo[c][l,2]oxaborol-3-yl)-acetic acid ethyl ester (2 g, 8 mmol), 2-chloro-4-nitro-pyridine oxide (2.2 g, 12 mmol) and CS2CO3 (5.7 g, 18 mmol) were mixed in DMF (50 mL) and stirred at roomtemperature for 1 h. Water was added and the reaction mixture was adjusted to pH 3. The reaction mixture was extracted with ethyl acetate. The organic layers were concentrated to give a residue. The residue was purified by flash columnchromatography (dichloromethane : methanol = 15 : 1) to give the title compound (0.77 g, 29%) as a brown solid. MS found (electrospray): (M + H)+ = 389.1. |
Reference:
[1]Current Patent Assignee: PFIZER INC - WO2011/17125, 2011, A1
Location in patent: Page/Page column 186-187
- 55
-
[ 14432-16-7 ]
-
 [ 1137826-05-1 ]
[ 1137826-05-1 ]
-
[ 1350750-78-5 ]
| Yield | Reaction Conditions | Operation in experiment |
|
|
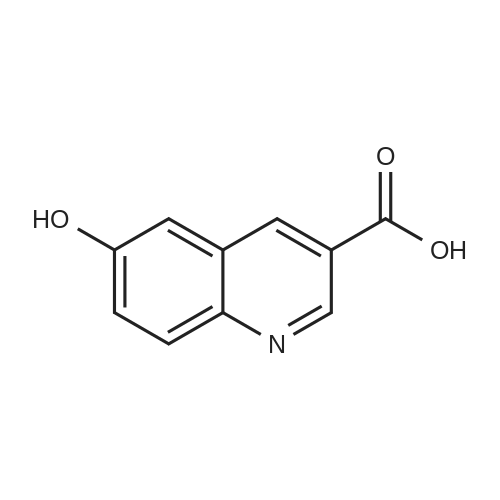
Example 20: -V-hydroxy-6-(pyridin-4-yloxy)quinoline-3-carboxamide Compound 1-32 lnt-33 i-32 Step 1: 6-(2-chloropyridin-4-yloxy)quinoline-3-carboxylic acid Interniediate-31 [00205] A mixture of 2-chloro-4-nitropyridine (0.228 g, 1.44 mmol), 6-hydroxyquinoline-3- carboxylic acid (Int-15, 0.3 g, 1.58 mmol), and cesium carbonate (1.41 g, 4.32 mol) in N,N- dimethylformamide (3 mL) was heated at 150 C for 2 h in a microwave reactor. The reaction was cooled to room temperature and filtered to remove solids. The filtrate was acidified with hydrochloric acid (1M in water) until pH 1.5. The product precipitated cleanly and was isolated by suction filtration. LC-MS:(FA) EA+ 301; NMR (400 MHz, rf6-DMSO) delta ppm 9.31 (d, J = 2.1 Hz, 1 H), 8.98 (d, J = 2.0 Hz, 1H), 8.34 (d, J = 5.7 Hz, 1 H), 8.21 (d, J = 9.1 Hz, 1 H), 8.06 (d, J = 2.7 Hz, 1 H), 7.79 (dd, J = 9.1, 2.8Hz, 1 H), 7.20 (d, J = 2.2 Hz, 1 H), 7.09 (dd, J = 5.7, 2.3 Hz, 1 H). |
Reference:
[1]Patent: WO2011/146591,2011,A1 .Location in patent: Page/Page column 77-78
- 56
-
[ 14432-16-7 ]
-
[ 291509-79-0 ]
-
[ 1231750-46-1 ]
| Yield | Reaction Conditions | Operation in experiment |
| 75% |
With potassium carbonate In N,N-dimethyl-formamide at 60℃; for 2h; |
51
Intermediate 517-methoxy-3-(4'-nitro-1'-oxy-3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-yl)-1.3.4,5-tetrahydro-benzo[d][1,3]diazepin-2-one; A mixture of 160 mg (0.581 mmol) 7-methoxy-3-piperidin-4-yl-1,3,4,5-tetrahydro-1,3-benzodiazepin-2-one, 100 mg (0.573 mmol) 2-chloro-4-nitro-pyridin-N-oxide and 100 mg (0.724 mmol) potassium carbonate in 2.0 mL DMF was stirred for 2 h at 60° C. until the reaction was complete. After cooling to RT the reaction mixture was poured onto water. The precipitate formed was filtered off, washed with water and dried i. vac.Yield: 180 mg (75% of theory)ESI-MS: m/z=414 (M+H)+ |
Reference:
[1]Current Patent Assignee: C.H. Boehringer Sohn AG & Co. KG - US2012/88755, 2012, A1
Location in patent: Page/Page column 82
- 57
-
[ 14432-16-7 ]
-
[ 108-91-8 ]
-
 [ 75291-50-8 ]
[ 75291-50-8 ]
| Yield | Reaction Conditions | Operation in experiment |
| 76% |
In ethanol at 80℃; for 13h; |
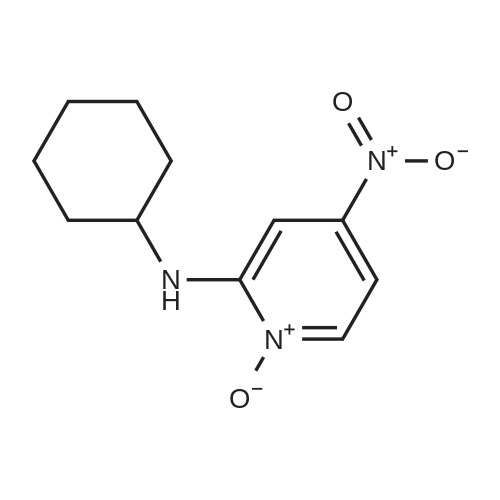
The mixture of 2-chloro-4-nitropyridine 1-oxide (1) (325 mg, 1.86 mmol) and cyclohexylamine (0.38 mL, 3.35 mmol) in EtOH (9 ml) was heated at 80 °C until compound 1 disappeared in TLC. After reaction termination, the mixture was cooled to ambient temperature and solvent was removed in vacuo. The concentrated crude product was purified by flash column chromatography with EA/Hex (1:1) as the eluent to produce cyclohexyl-(4-nitro-1-oxy-pyridin-2-yl)-amine (2a) as a yellow solid (76%); 1H NMR (400 MHz, CDCl3) δ 8.23 (1H, br, s), 7.39 (1H, s), 7.36 (1H, br, s), 6.92 (1H, br, s), 3.44 (1H, br, s), 2.04-2.06 (2H, m), 1.72-1.90 (2H, m), 1.56-1.60 (1H, m), 1.42-1.47 (3H, m), 1.26-1.30 (2H, m). |
Reference:
[1]Kim, Mi-Hyun; Lee, Junghun; Jung, Kyungjin; Kim, Minjung; Park, Yun-Jin; Ahn, Heechul; Kwon, Young Hye; Hah, Jung-Mi
[Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 8, p. 2271 - 2285]
- 58
-
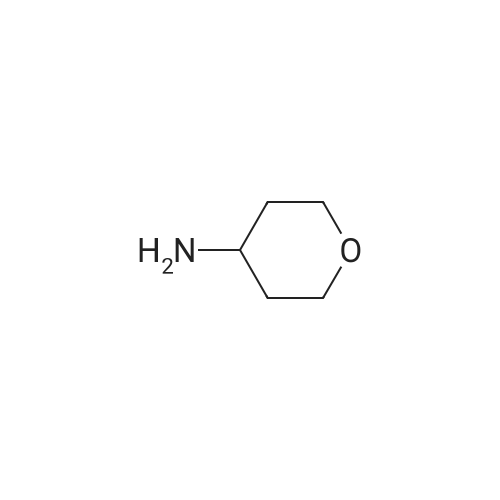 [ 38041-19-9 ]
[ 38041-19-9 ]
-
[ 14432-16-7 ]
-
[ 1428958-49-9 ]
| Yield | Reaction Conditions | Operation in experiment |
| 51% |
In ethanol at 80℃; for 13h; |
General procedure: The mixture of 2-chloro-4-nitropyridine 1-oxide (1) (325 mg, 1.86 mmol) and cyclohexylamine (0.38 mL, 3.35 mmol) in EtOH (9 ml) was heated at 80 °C until compound 1 disappeared in TLC. After reaction termination, the mixture was cooled to ambient temperature and solvent was removed in vacuo. The concentrated crude product was purified by flash column chromatography with EA/Hex (1:1) as the eluent to produce cyclohexyl-(4-nitro-1-oxy-pyridin-2-yl)-amine (2a) as a yellow solid (76%). |
Reference:
[1]Kim, Mi-Hyun; Lee, Junghun; Jung, Kyungjin; Kim, Minjung; Park, Yun-Jin; Ahn, Heechul; Kwon, Young Hye; Hah, Jung-Mi
[Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 8, p. 2271 - 2285]
- 59
-
[ 14432-16-7 ]
-
1-amino-propan-2-ol
[ No CAS ]
-
1-(4-nitro-1-oxy-pyridin-2-ylamino)-propan-2-ol
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 99% |
In ethanol at 80℃; for 13h; |
General procedure: The mixture of 2-chloro-4-nitropyridine 1-oxide (1) (325 mg, 1.86 mmol) and cyclohexylamine (0.38 mL, 3.35 mmol) in EtOH (9 ml) was heated at 80 °C until compound 1 disappeared in TLC. After reaction termination, the mixture was cooled to ambient temperature and solvent was removed in vacuo. The concentrated crude product was purified by flash column chromatography with EA/Hex (1:1) as the eluent to produce cyclohexyl-(4-nitro-1-oxy-pyridin-2-yl)-amine (2a) as a yellow solid (76%). |
Reference:
[1]Kim, Mi-Hyun; Lee, Junghun; Jung, Kyungjin; Kim, Minjung; Park, Yun-Jin; Ahn, Heechul; Kwon, Young Hye; Hah, Jung-Mi
[Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 8, p. 2271 - 2285]
- 60
-
[ 14432-16-7 ]
-
tert-butyl 3-aminopiperidine-1-carboxylate
[ No CAS ]
-
3-(4-nitro-1-oxy-pyridin-2-ylamino)-piperidine-1-carboxylic acid tert-butyl ester
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 67% |
In ethanol at 80℃; for 13h; |
General procedure: The mixture of 2-chloro-4-nitropyridine 1-oxide (1) (325 mg, 1.86 mmol) and cyclohexylamine (0.38 mL, 3.35 mmol) in EtOH (9 ml) was heated at 80 °C until compound 1 disappeared in TLC. After reaction termination, the mixture was cooled to ambient temperature and solvent was removed in vacuo. The concentrated crude product was purified by flash column chromatography with EA/Hex (1:1) as the eluent to produce cyclohexyl-(4-nitro-1-oxy-pyridin-2-yl)-amine (2a) as a yellow solid (76%). |
Reference:
[1]Kim, Mi-Hyun; Lee, Junghun; Jung, Kyungjin; Kim, Minjung; Park, Yun-Jin; Ahn, Heechul; Kwon, Young Hye; Hah, Jung-Mi
[Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 8, p. 2271 - 2285]
- 61
-
[ 14432-16-7 ]
-
[ 124-40-3 ]
-
[ 36100-42-2 ]
| Yield | Reaction Conditions | Operation in experiment |
| 91% |
In tetrahydrofuran; ethanol at 90℃; for 5h; |
2-(dimethylamino)-4-nitropyridine 1 -oxide (i50):
2-(dimethylamino)-4-nitropyridine 1 -oxide (i50): To a stirred solution of 2-chloro-4-nitropyridine 1 -oxide (i49) (2.1 g, 12 mmol) in THF: Ethanol 1 :1 (50 mL), a 2 M dimethylamine solution in THF (1 .24 g, 27.6 mmol) was added drop wise and the reaction was heated at 90°C for 5h. The progress of the reaction was monitored by TLC. After completion, the mixture was concentrated under reduced pressure. The crude product was purified by silica gel (100:200 mesh) column chromatography using 2% methanol in dichloromethane as eluent to afford 2-(dimethylamino)-4-nitropyridine 1 -oxide (i50) (2.0 g, 91 %). 1H NMR (400 MHz, Chloroform-d) δ 3.1 1 (s, 6H), 7.64 (m, 2H), 8.21 (d, J = 7.0 Hz, 1 H). MS (ESI) m/e (M+1 )+: 184 |
|
In water at 100℃; Inert atmosphere; Sealed tube; |
|
Reference:
[1]Current Patent Assignee: UCB - WO2016/124508, 2016, A1
Location in patent: Page/Page column 66
[2]Mita, Tsuyoshi; Michigami, Kenichi; Sato, Yoshihiro
[Chemistry - An Asian Journal, 2013, vol. 8, # 12, p. 2970 - 2973]
- 63
-
[ 14432-16-7 ]
-
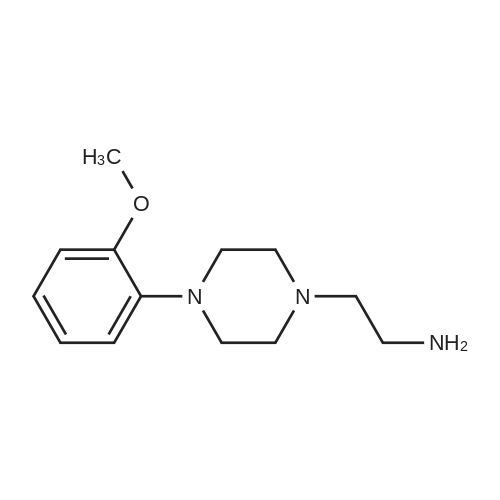 [ 40255-48-9 ]
[ 40255-48-9 ]
-
N-{2-[4-(2-methoxyphenyl)piperazin-1-yl]-ethyl}-N-(4-nitro-1-oxypyridin-2-yl)amine
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 31% |
In ethanol at 100℃; for 1h; |
N-{2-[4-(2-methoxy-phenyl)-piperazin-1-yl]-ethyl}-N-(4-nitro-1-oxy-pyridin-2-yl)-amine (5)
In a 5 mL vial flask from Biotage Initiator microwave synthesizer 2-[4-(2-methoxy-phenyl)-piperazin-1-yl]-ethylamine 3 (240 mg, 1.02 mmol), 2-chloro-4-nitropyridine-N-oxide 4 (267 mg, 1.53 mmol) and EtOH (4 mL) were mixed. The vial was capped and heated up to 100 C for 60 min. Then the solvent was removed under reduced pressure and the residue dissolved in CH2Cl2 (8 mL) and treated with an excess of solid potassium carbonate to neutralize the HCl evolved from the reaction. The solid was filtered off and the filtrate was evaporated. The residue was purified by silica gel flash chromatography using a gradient of AcOEt/MeOH (1:0-5:1) as eluent to give a yellow solid (118 mg; 31%). m. p. 135-137C. 1H-NMR (300 MHz, CDCl3): 8.20 (1H, d, J = 5.6 Hz, H6py), 7.44 (1H, dd, J = 2.0 Hz, J = 5.6 Hz, H5py), 7.37 (1H, d, J = 2.0 Hz, H3py), 7.32 (1H, br. s, NH), 7.01-6.83 (4H, m, HAr), 3.84 (3H, s, OCH3), 3.43 (2H, ap. c, J = 5.3 Hz, J = 6.3 Hz, CH2NH), 3.10 (4H, br. s, 2 x CH2N), 2.78 (2H, t, J = 6.3 Hz, CH2N), 2.71 (4H, m, 2 x CH2N) ppm. 13C-NMR (50 MHz, CDCl3): 152.1, 150.6, 144.6, 141.0, 137.6, 122.9, 120.9, 118.2. 111.0, 105.4, 99.3, 56.1, 55.3, 53.1, 50.5, 39.1 ppm. IR (max, NaCl): 3328, 2939, 2820, 1628, 1541, 1500, 1451, 1345, 1306, 1240, 1026, 740, 658 cm-1. HRMS (ESI-TOF): Calculated for C18H24N5O4 [M+H]+: 374.1828, found 374.1830. |
Reference:
[1]García, Gonzalo; Abet, Valentina; Alajarín, Ramón; Álvarez-Builla, Julio; Delgado, Mercedes; García-García, Luis; Bascuñana-Almarcha, Pablo; Peña-Salcedo, Carmen; Kelly, James; Pozo, Miguel A.
[European Journal of Medicinal Chemistry, 2014, vol. 85, p. 795 - 806]
- 68
-
3-hydroxyl-3-methylazetidine hydrochloride
[ No CAS ]
-
[ 14432-16-7 ]
-
2-(3-hydroxy-3-methylazetidin-1-yl)-4-nitropyridine-1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With caesium carbonate In acetonitrile for 1h; Reflux; |
94.1 Step 1 Preparation of 2-(3-hydroxy-3-methylazetidin-1-yl)-4-nitropyridine-1-oxide
Dissolve 2-chloro-4-nitropyridine-N-oxide (0.2g, 1.14mmol) in5mL of acetonitrile solution, then add 3-methylazetidine-3-ol hydrochloride to the reaction system(0.35g, 2.8mmol) and cesium carbonate (1.3g, 3.99mmol), and then stirred under reflux for 1h.The reaction is complete,Cool to room temperature. Filter with suction, wash the filter cake with acetonitrile, collect the filtrate, and concentrate under reduced pressure to obtain the title compound. |
Reference:
[1]Current Patent Assignee: NANJING SANHOME INVESTMENT GROUP CO LTD - CN111196804, 2020, A
Location in patent: Paragraph 0941; 0943-0945
- 69
-
[ 14432-16-7 ]
-
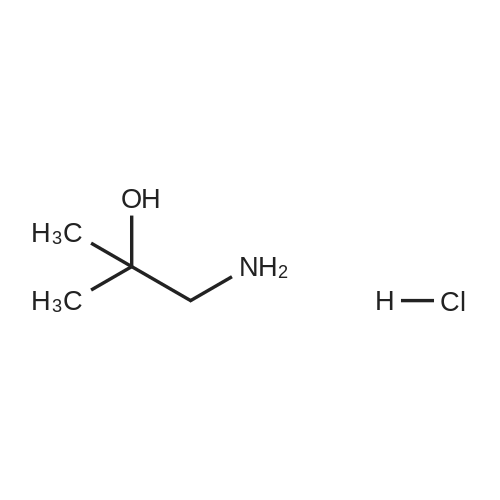 [ 30533-50-7 ]
[ 30533-50-7 ]
-
2-((2-hydroxy-2-methylpropyl)amino)-4-nitropyridine-1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
With carbonic-acid In acetonitrile at 90℃; for 4.5h; Inert atmosphere; |
93.1 Step 1 Preparation of 2-((2-hydroxy-2-methylpropyl)amino)-4-nitropyridine-1-oxide
Place 2-chloro-4-nitropyridine-1-oxide in a 50mL single-necked bottle(0.2g, 1.15mmol) dissolved in acetonitrile(20mL), add 1-amino-2-methyl-2-propanol hydrochloride(0.12g, 1.26mmol)withCarbonic acid(1.12g, 3.44mmol). Under nitrogen protection, the reaction was refluxed at 90 for 4.5h.After the reaction,Add water and ethyl acetate, extract, combine the organic phase, and wash the organic phase with saturated brine,Dry over anhydrous sodium sulfate, concentrate under reduced pressure, and purify by column chromatography to obtain the title compound. |
Reference:
[1]Current Patent Assignee: NANJING SANHOME INVESTMENT GROUP CO LTD - CN111196804, 2020, A
Location in patent: Paragraph 0930; 0932-0934
- 70
-
[ 14432-16-7 ]
-
(S)-2-(2-((2,6-diisopropylphenyl)carbamoyl)pyrrolidin-1-yl)-4-(pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 71
-
[ 14432-16-7 ]
-
(S)-4-chloro-2-(2-((2,6-diisopropylphenyl)carbamoyl)pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: acetyl chloride / 4 h / 70 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 72
-
[ 14432-16-7 ]
-
(S)-2-(2-((2,6-diisopropylphenyl)carbamoyl)pyrrolidin-1-yl)-4-(dimethylamino)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 3 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: acetyl chloride / 4 h / 70 °C
3: water / 4 h / 100 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 73
-
[ 14432-16-7 ]
-
(S)-N-(2,6-diisopropylphenyl)-1-(4-(pyrrolidin-1-yl)pyridin-2-yl)pyrrolidine-2-carboxamide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 3 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C
3: hydrogen; palladium 10% on activated carbon / methanol / 48 h / 20 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 74
-
[ 14432-16-7 ]
-
(2S)-N-phenylpyrrolidine-2-carboxamide
[ No CAS ]
-
(S)-2-(2-(phenylcarbamoyl)pyrrolidin-1-yl)-4-(pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 75
-
[ 14432-16-7 ]
-
(2S)-N-phenylpyrrolidine-2-carboxamide
[ No CAS ]
-
(S)-4-nitro-2-(2-(phenylcarbamoyl)pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 86% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 76
-
(S)-N-(2,6-diethylphenyl)pyrrolidine-2-carboxamide
[ No CAS ]
-
[ 14432-16-7 ]
-
(S)-2-(2-((2,6-diethylphenyl)carbamoyl)pyrrolidin-1-yl)-4-(pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 77
-
(S)-N-(2,6-diethylphenyl)pyrrolidine-2-carboxamide
[ No CAS ]
-
[ 14432-16-7 ]
-
(S)-2-(2-((2,6-diethylphenyl)carbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 85% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 78
-
[ 943784-90-5 ]
-
[ 14432-16-7 ]
-
(S)-2-(2-((2,6-diisopropylphenyl)carbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 90% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 79
-
[ 14432-16-7 ]
-
[ 748149-61-3 ]
-
(S)-2-(2-(benzhydrylcarbamoyl)pyrrolidin-1-yl)-4-(pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 80
-
[ 14432-16-7 ]
-
[ 748149-61-3 ]
-
(S)-2-(2-(benzhydrylcarbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 88% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 81
-
[ 14432-16-7 ]
-
[ 875542-94-2 ]
-
(S)-2-(2-((3,5-bis(trifluoromethyl)phenyl)carbamoyl)pyrrolidin-1-yl)-4-(pyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
|
Multi-step reaction with 2 steps
1: triethylamine / tetrahydrofuran / 10 h / 70 °C
2: 8 h / 80 °C |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 82
-
[ 14432-16-7 ]
-
[ 875542-94-2 ]
-
(S)-2-(2-((3,5-bis(trifluoromethyl)phenyl)carbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 78% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 83
-
[ 14432-16-7 ]
-
[ 128018-18-8 ]
-
(S)-2-(2-(tert-butylcarbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 82% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 84
-
[ 14432-16-7 ]
-
[ 1173169-85-1 ]
-
(S)-2-(2-(methyl(phenyl)carbamoyl)pyrrolidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 70% |
With triethylamine In tetrahydrofuran at 70℃; for 10h; |
|
Reference:
[1]Deng, Yun; Guo, Hai-Ming; Huang, Bin; Li, Ning; Qu, Gui-Rong; Tian, Yin; Wu, Xiao-Xia; Xie, Ming-Sheng
[Journal of the American Chemical Society, 2020, vol. 142, # 45, p. 19226 - 19238]
- 85
-
[ 14432-16-7 ]
-
2-(azetidin-3-yl)propan-2-ol
[ No CAS ]
-
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
- 86
-
[ 14432-16-7 ]
-
 [ 848392-24-5 ]
[ 848392-24-5 ]
-
2-(3-hydroxy-3-(trifluoromethyl)azetidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
- 87
-
3,3-difluoroazetidine
[ No CAS ]
-
[ 14432-16-7 ]
-
2-(3,3-difluoroazetidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 50% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 50% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
- 88
-
 [ 732976-86-2 ]
[ 732976-86-2 ]
-
[ 14432-16-7 ]
-
2-(3-cyanoazetidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 70% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
- 89
-
[ 14432-16-7 ]
-
 [ 936909-11-4 ]
[ 936909-11-4 ]
-
2-(3-cyano-3-methylazetidin-1-yl)-4-nitropyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 75% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 75% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
- 90
-
 [ 1841-00-5 ]
[ 1841-00-5 ]
-
[ 14432-16-7 ]
-
4-nitro-2-(3,3,4,4-tetrafluoropyrrolidin-1-yl)pyridine 1-oxide
[ No CAS ]
| Yield | Reaction Conditions | Operation in experiment |
| 59% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
| 59% |
With Cs2CO3 In acetonitrile at 60℃; Reflux; Inert atmosphere; |
2-(3-(2-hydroxypropan-2-yl)azetidin-1-yl)-4-nitropyridine 1-oxide (26)
General procedure: A solution of 25 (0.52 g4.54 mmol), 2-chloro-4-nitropyridine 1-oxide (0.4 g, 2.28 mol) and Cs2CO3 (2.23 g, 6.86 mmol) in acetonitrile (20 mL) was heated to reflux overnight at 60 °C under nitrogen. After completion, the solution was concentrated and the residue was purified by silica gel column chromatography to afford 26 (0. 81 g, 70%). |
Reference:
[1]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
[2]Wang, Hai; Wang, Xiaowei; Wang, Zhenwei; Yu, Zhuangzhuang; Zhang, Yan; Zhao, Liwen
[Bioorganic and Medicinal Chemistry Letters, 2022, vol. 61]
 630-580-1088
630-580-1088
 sales@ambeed.com
sales@ambeed.com
 Hello, Sign in
Hello, Sign in

 0
0

 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science
 Contact Us
Contact Us
 0
0



 HazMat Fee +
HazMat Fee +
 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping