一种基于三代测序检测多样本量比较转录组分析方法与流程
本发明属于生物信息学技术领域,尤其涉及一种基于三代测序检测多样本量比较转录组分析方法。
背景技术:
随着测序技术的飞速发展,许多基因组已经完成组装,比较基因组学也成为了研究这些物种与近缘物种进化关系、查找与物种间进化相关基因的重要手段。但相对于已完成基因组测序的物种,未完成基因组测序的物种依然占据绝大多数。这有两方面的原因,一是基于目前的测序技术,组装一个基因组的费用依然十分昂贵;二是某些物种的基因组比较复杂,如大基因组物种、多倍体物种等,组装难度很大,目前的组装技术很难实现。对于这些物种,可以用比较转录组的方法,利用它们的转录组数据开展相应的研究,该方法已经在一些科属中成功运用。全长转录组是指基于三代测序技术测出的转录组,主要有以下优点:(1)可以直接获得全长转录本;(2)三代全长无gc偏好性,无需组装,结果更准确;(3)数据准确性高,覆盖更加全面。
随着测序费用的逐年下降,越来越多的科研工作者倾向于多样本量的测序分析,但是目前基于比较转录组的分析方法还比较少见。
技术实现要素:
为克服上述现有问题或者至少部分地解决上述问题,本发明实施例提供一种基于三代测序检测多样本量比较转录组分析方法。
本发明实施例提供了一种基于三代测序检测多样本量比较转录组分析方法,包括:
三代测序下机数据质控,得到质控数据结果;
基于所述质控数据结果,获取三代全长转录本,对所述三代全长转录本进行cds和pep预测,得到预测结果;
基于预测结果进行基因家族聚类,获取直系同源基因、旁系同源基因和物种特有基因;
对所述直系同源基因进行宏观进化分析、ssr检测和标记开发,以及;
对所述直系同源基因进行功能基因定位,挖掘研究材料不同亚种或近源物种快速进化基因;
对所述旁系同源基因进行wgd事件检测和个别家族的基因家族树分析;
对所述物种特有基因进行数据库注释,明确研究材料的特有基因的特有功能。
在上述技术方案的基础上,本发明实施例还可以作出如下改进。
可选的,所述三代测序下机数据质控,得到质控数据结果包括:
针对nanopore平台测序数据进行过滤接头,过滤掉质量值小于7的reads,且过滤掉长度小于300bp的reads;
针对pacbio平台测序数据进行过滤接头,过滤掉长度小于300bp的reads。
可选的,所述基于所述质控数据结果,获取三代全长转录本,对所述三代全长转录本进行cds和pep预测,得到预测结果包括:
针对nanopore平台测序数据,基于对应的质控数据结果,采用pychopper中的cdna_classifier.py程序进行全长转录本检测,同时利用pinfish针对获取的全长转录本进行自纠错,进而获得高质量的全长转录本;
针对pacbio平台测序数据,基于对应的质控数据结果,采用软件smrtlink7.0,结合参数minpredictedaccuracy和hp_min_accuracy进行分析,获取质量值在0.99以上的高质量全长转录本;
基于所述全长转录本,利用transdecoder-3.0.1进行cds和pep预测,每个全长转录本预测多个开放阅读框orf,选择最长的orf作为全长转录本的预测结果。
可选的,所述基于预测结果进行基因家族聚类,获取直系同源基因、旁系同源基因和物种特有基因包括:
预测的pep信息,利用blastp程序进行all-vs-all最佳互惠比对,以e-value为1e-10进行搜索,选择besthitstop5%;采用最小二乘法将top5%的分数值拟合成一个线性模型,利用比特值进行模型转换;基于pep长度和系统发育距离对blastp比特值得分进行标准化,使用rbnh方法,确定同源组序列性相似度的阈值线,构建出高质量的直系同源组图;
基于直系同源组图,构建直系同源正交组,通过归一化的比特值画出正交组的边缘连接,作为mcl的输入文件;
基于mcl,将直系同源正交组进行聚类,基于聚类结果判断研究材料为直系同源基因或特有基因家族或共有基因家族或旁系同源基因;
其中,所述基于聚类结果判断研究材料为直系同源基因或特有基因家族或共有基因家族或旁系同源基因包括:
如果在某一个基因家族,所有研究材料只有一个单拷贝基因,将所有研究材料定义为直系同源基因;
如果某基因家族只有一个研究材料的基因,将研究材料定义为特有基因家族;
如果所有研究材料在某个基因家族至少包含一个基因,将研究材料定义为共有基因家族;
针对某个研究材料,如果在某个基因家族有多个基因,将该研究材料定义为旁系同源基因。
可选的,对所述直系同源基因进行宏观进化分析包括:
根据直系同源基因,判定单拷贝基因的数目;若单拷贝基因的数目至少300个,则直接利用单拷贝基因家族构建系统发育树;其中,所述直接利用单拷贝基因家族构建物种系统发育树包括:
利用对应的cds预测信息和pep预测信息进行超矩阵法分别构建物种系统发育树;或者,针对每个基因分别构建基因树,利用astral进行溯祖法结合单拷贝基因树构建物种系统发育树;
如果若单拷贝基因的数目少于300个,则为每个直系同源组构建基因系统发育树,使用stag算法从无根基因树上构建无根物种树,使用stride算法构建有根物种树,有根物种树进一步辅助构建有根基因树;
基于构建的物种系统发育树,结合提供的化石证据信息进行分化时间的分析,包括:
以某一特定研究材料的化石记录作为参照点,通过基因序列间的分歧程度以及基因序列的平均置换速率来估计速率恒定分支间的分歧时间,同时计算物种系统发育树上其它节点的发生时间,推测相关研究材料的起源时间和不同研究材料的分歧时间。
可选的,对所述直系同源基因进行ssr检测和标记开发包括:
针对单拷贝直系同源基因,利用primer3进行ssr检测分析,挑选物种保守的多态ssr用于物种或近源种的区分。
可选的,对所述直系同源基因进行功能基因定位,挖掘研究材料不同亚种或近源物种快速进化基因包括:
采用预设模型对所述直系同源基因进行选择压力分析;
其中,所述预设模型为分支模型,所述分支模型对基因系统发育树中的不同枝系的w值的异质性进行界定,界定结果包括基因系统发育树中所有枝系的w值相等,基因系统发育树中所有枝系的w值不相等及基因系统发育树中前景枝和背景枝的w值不同;
所述预设模型为位点模型,所述位点模型对基因系统发育树的不同分支所受选择压力相同、但不同的氨基酸位点的选择压力进行分析;
所述预设模型为枝位点模型,当定位点间的w值变化,且支系间的w值变化时,所述枝位点模型用于分析前景枝中正选择作用对部分位点的影响。
可选的,对所述物种特有基因进行数据库注释,明确研究材料的特有基因的特有功能包括:
提取每个材料在基因家族的特有基因,利用blast进行go、cog、kegg、pfam、swissprot数据库进行序列相似性比对,获取物种的特有基因功能。
可选的,对所述旁系同源基因进行wgd事件检测包括:
针对某个研究材料潜在发生的wgd事件,提取研究材料在基因家族中呈现多拷贝的基因;
进行两两基因的全局比对,剔除比对结果中gap区域超过50%的基因对;
分析高质量比对的基因对之间的同义替代率ks值,如果在某基因家族只有一个基因对,则计算的ks值代表该基因家族;如果有多个基因对,则计算ks值的中位数代表该基因家族;
采用高斯正态模型拟合所有基因家族的ks值,得到最高峰的ks值,根据公式t=ks/2μ计算wgd的时间,其中μ代表中性突变率,t为基因发生时间。
本发明实施例提供一种基于三代测序检测多样本量比较转录组分析方法,该方法在处理多样本量比较转录组数据,针对检测的全长转录本进行cds/pep预测;随后进行宏观进化分析,主要包括系统发育分析和分化时间分析;针对预测的单拷贝直系同源基因进行ssr检测,标记开发;针对单拷贝直系同源基因进行选择压分析,挖掘物种适应性进化和快速进化基因;针对旁系同源基因个别家族进行基因家族树分析;针对特殊材料进行wgd事件分析,整体上,提供了一套完善的科学研究方法进行生物信息学的研究。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例提供的基于三代测序检测多样本量比较转录组分析方法整体流程示意图;
图2为本发明实施例的reads测序循环数与质量值分布图;
图3为本发明实施例的8个样品的系统发育树图形;
图4为本发明实施例的8个样品的分化时间图形。
具体实施方式
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
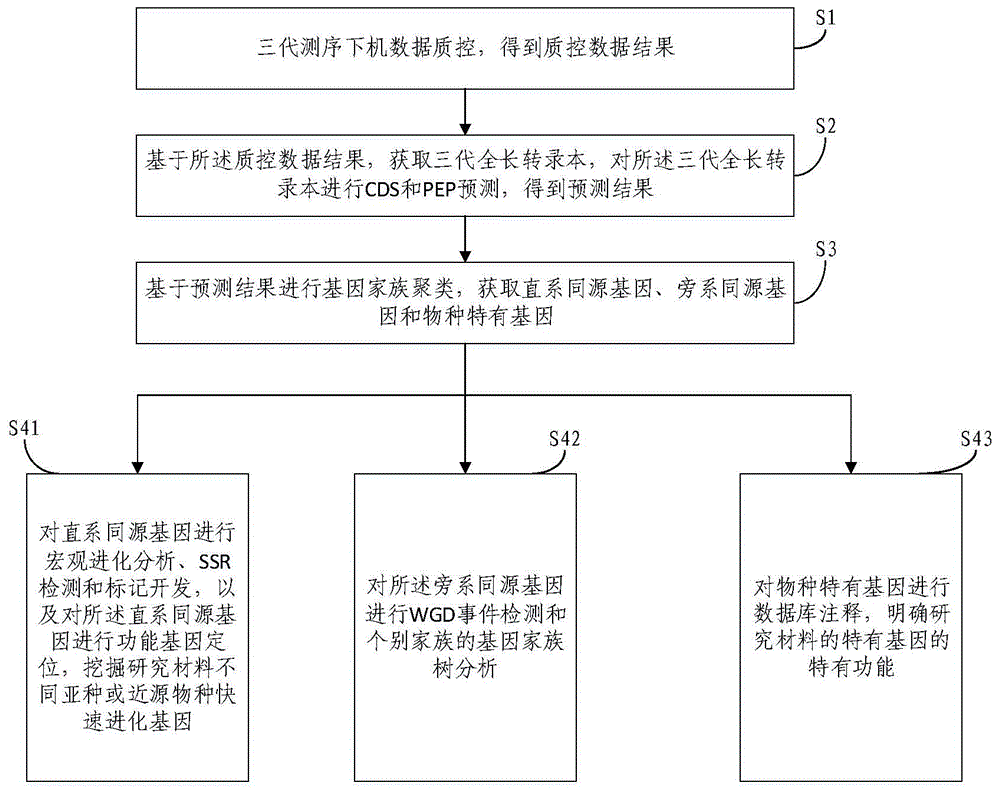
在本发明的一个实施例中提供一种基于三代测序检测多样本量比较转录组分析方法,图1为本发明实施例提供的基于三代测序检测多样本量比较转录组分析方法整体流程示意图,该方法包括:
三代测序下机数据质控,得到质控数据结果;
基于所述质控数据结果,获取三代全长转录本,对所述三代全长转录本进行cds和pep预测,得到预测结果;
基于预测结果进行基因家族聚类,获取直系同源基因、旁系同源基因和物种特有基因;
对所述直系同源基因进行宏观进化分析、ssr检测和标记开发,以及;
对所述直系同源基因进行功能基因定位,挖掘研究材料不同亚种或近源物种快速进化基因;
对所述旁系同源基因进行wgd事件检测和个别家族的基因家族树分析;
对所述物种特有基因进行数据库注释,明确研究材料的特有基因的特有功能。
可以理解的是,本发明实施例在处理多样本量比较转录组数据时,针对检测的全长转录本进行cds/pep预测;随后进行宏观进化分析,主要包括系统发育分析和分化时间分析;针对预测的单拷贝直系同源基因进行ssr检测,标记开发;针对单拷贝直系同源基因进行选择压分析,挖掘物种适应性进化和快速进化基因;针对旁系同源基因个别家族进行基因家族树分析;针对特殊材料进行wgd事件分析。整体上,提供了一套完善的科学研究方法进行生物信息学的研究。
作为一个可选的实施例,本发明实施例中,三代测序下机数据质控,得到质控数据结果包括:
针对nanopore三代测序下机数据,利用porechop过滤接头,根据下机数据特点保留质量值大于等于7,且片段长度不小于300bp的reads;
针对pacbio三代测序下机数据,过滤接头,过滤片段长度小于300bp的reads,至于质量值,暂时先不考虑。
作为一个可选的实施例,基于质控数据结果,检测获取三代全长转录本的步骤为:
对nanopore平台测序数据,采用pychopper中的cdna_classifier.py程序进行全长转录本检测,获得初始的三代全长reads信息。由于三代测序中碱基的高错误率(平均在10-15%之间),利用pinfish相关程序针对获取的全长转录本进行自纠错,进而获得高质量的全长转录本。首先利用minimap2将三代全长转录本比对到参考基因组上,获得比对结果文件bam,随后利用spliced_bam2gff将bam文件转换为gff2格式,记录每一个特定的转录本。利用cluster_gff将gff2文件中具有相似外显子/内含子结构的转录本聚到一起,并根据聚类结果获取外显子的边界从而构建consensus(一致性)序列。根据cluster_gff程序定义的一致性序列利用racon进行纠错,从而得到高质量的全长转录本。
针对pacbio平台测序数据,采用软件smrtlink7.0,首先利用ccs针对质控数据获取高度一致的consensus序列,从图2中可以看出,随着测序reads的pass(循环数)越多,consensus的phread(质量值)越高。利用smrtlink7.0中的isoseq3中的cluster将ccs进行聚类,提取全长转录本和非全长转录本,然后利用polish程序根据非全长转录本校正全长转录本,最后利用参数hp_min_accuracy(0.99)全长转录本分为高质量全长转录本(>=0.99)和低质量全长转录本,高质量全长转录本参与下游的基因家族聚类分析。
作为一个可选的实施例,对三代全长转录本进行cds和pep预测,得到预测结果的步骤为:
无论基于nanopore测序,亦或是pacbio测序,均是基于上述实施例获取的高质量全长转录本进行cds和pep的检测,具体检测步骤为,利用transdecoder-3.0.1对全长转录本识别长度至少为100个氨基酸的orf(开放阅读框),同时利用blast比对nr以及swissprot蛋白数据库,然后提取比对上的同源序列的位置识别orf,结果存储到后缀名分别是pep、cds、gff3的文件中。理论上每个基因可以有不同的可变剪接,即每个基因会有多个转录本,选择最长的orf(>=300bp)代表该基因。
作为一个可选的实施例,所述基于预测结果进行基因家族聚类,获取直系同源基因、旁系同源基因和物种特有基因包括:
预测的pep信息,利用blastp程序进行all-vs-all最佳互惠比对,以e-value为1e-10进行搜索,选择besthitstop5%;采用最小二乘法将top5%的分数值拟合成一个线性模型,利用比特值进行模型转换;基于pep长度和系统发育距离对blastp比特值得分进行标准化,使用rbnh方法,确定同源组序列性相似度的阈值线,构建出高质量的直系同源组图;
基于直系同源组图,构建直系同源正交组,通过归一化的比特值画出正交组的边缘连接,作为mcl的输入文件;
基于mcl,将直系同源正交组进行聚类,基于聚类结果判断研究材料为直系同源基因或特有基因家族或共有基因家族或旁系同源基因;
其中,所述基于聚类结果判断研究材料为直系同源基因或特有基因家族或共有基因家族或旁系同源基因包括:
如果在某一个基因家族,所有研究材料只有一个单拷贝基因,将所有研究材料定义为直系同源基因;
如果某基因家族只有一个研究材料的基因,将研究材料定义为特有基因家族;
如果所有研究材料在某个基因家族至少包含一个基因,将研究材料定义为共有基因家族;
针对某个研究材料,如果在某个基因家族有多个基因,将该研究材料定义为旁系同源基因。
作为一个可选的实施例,对直系同源基因进行宏观进化分析主要包括系统进化和分化时间分析,具体包括以下步骤:
首先基于研究材料判定单拷贝基因的数目,如果此种类型的基因家族数目不小于300个,则可以直接利用单拷贝基因家族进行系统发育树构建。可进一步细化为:(1),分别针对单拷贝基因家族的cds和pep序列进行多样本全局比对,序列对齐后,剔除比对结果中gap区域超过50%的基因对,然后将所有单拷贝基因家族的比对结果进行串联,构建两个超大矩阵(分别是cds和pep序列),然后基于两个超大矩阵构建系统发育树;(2),分别针对单拷贝基因家具的pep序列进行多样本全局比对,同样的,序列对齐后剔除gap区域超过50%的基因对,这样得到众多的全局比对结果(不少于300个),每种基因比对结果都构建基因系统发育树。随后利用astral进行溯祖法构建物种系统发育树,使用stag算法从无根基因树上构建无根物种树,使用stride算法构建有根物种树,有根物种树进一步辅助构建有根基因树。
根据上述步骤中构建的物种系统发育树,结合提供的化石证据信息进行分化时间的分析。以某一特定材料的化石记录作为参照点,通过基因序列间的分歧程度以及序列的平均置换速率(分子钟)来估计速率恒定分支间的分歧时间,同时计算系统树上其它节点的发生时间,从而推测相关研究材料的起源时间和不同材料的分歧时间。
作为一个可选的实施例,对所述直系同源基因进行ssr检测和标记开发包括:
根据基因家族聚类获取的单拷贝基因家族,利用primer3进行ssr分析,挑选物种保守的多态ssr进行引物设计,每种ssr根据基因序列设计2-3对,用于物种或近源种的区分。
作为一个可选的实施例,基于直系同源基因进行选择压力分析,挖掘研究材料不同亚种或近源物种快速进化基因。其中,针对特定物种之间的选择压力分析,主要包括以下几个模型:分支模型(branchmodel),主要是对基因系统发育树中的不同枝系(lineage)的w值的异质性进行界定,主要model有:one-ratiomodel,即系统发育树中所有w值是相等的;free-ratiomodel,即系统发育树中所有枝系的w值不相等;two-ratiomodel,即系统发育树中前景枝和背景枝的w值不同。针对研究材料,一般设置two-ratiomodel和one-ratiomodel进行似然比检验,如果w=1,即为中性进化,基因不受选择;如果w>1,基因受到正选择;如果w<1,基因受到纯化选择。
位点模型(sitemodel),同样是对基因系统发育树的不同分支所受选择压力相同,但不同的氨基酸位点经历的选择压力各异。这里主要有7个不同的假设模型,分别是:(1)m0(单一比率),即假设所有位点具有相同的w值;(2)m1a(近中性),假设仅有保守位点(0<w<1)和中性位点(w=1)而没有正选择位点(w>1)存在,这两类位点的比率分别为p0和p1,其对应的w值分别为w0和w1;(3)m2a,该模型在m1a基础上增加了第三类w值,即假设保守位点和中性位点外,存在处于正选择压力下的位点(w>1),这三类位点的比率分别为p0、p1和p2,其对应的w值分别为w0、w1和w2;(4)m3(离散),假设所有的位点w值呈简单的离散分布趋势;(5)m7(beta),假设所有的位点w属于矩阵(0,1)并呈beta分布;(6)m8(beta&w),该模型在m7基础上增加另一类w值(w>1);(7)m8a(beta&w>1),与m8模型类似,但将w值固定为1(w=0)。
枝位点模型(branch-sitemodel),假定位点间的w值是变化的,同时也假定支系间的w值是变化的,该模型主要用于前景枝中正选择作用对部分位点的影响。
作为一个可选的实施例,对所述物种特有基因进行数据库注释,明确研究材料的特有基因的特有功能包括:
提取每个材料在基因家族的特有基因,利用blast进行go、cog、kegg、pfam、swissprot数据库进行序列相似性比对,获取物种的特有基因功能。
作为一个可选的实施例,对旁系同源基因进行wgd事件检测的步骤为:
wgd是生物进化的主要因素之一,目前主要有两种方法进行检测:共线性分析和ks分布图。ks定义为平均每个同义替换位点上的同义置换数。如果研究材料没有wgd或者大片段重复,基因组中的旁系同源基因的同义置换符合指数分布,反之ks分布图中就会出现一个由于wgd导致的正态分布峰,而古老的wgd年龄则可通过分析这些峰中的同义置换数目进行预测。针对研究的某个材料潜在发生的wgd事件,提取研究材料在基因家族中呈现多拷贝(>=2)的基因。然后进行两两基因的全局比对,剔除比对结果中gap区域超过50%的基因对,随后分析高质量比对的基因对之间的ks值。如果在某基因家族只有一个基因对,则计算的ks值代表该基因家族,如果有多个基因对,则计算ks值的中位数代表该基因家族。采用高斯正态模型拟合所有基因家族的ks结果,得到符合正态分布峰值的ks值,根据公式t=ks/2μ计算wgd的时间,其中μ代表中性突变率,t即事件发生时间。
作为一个可选的实施例,基于旁系同源基因进行个别家族的基因家族树分析的目的为,基因家族的基因在物种之间都是比较保守的,通过对该家族多物种构建系统发育树,从而得到物种起源进化或亲缘关系方面的信息,同时挖掘物种中哪些基因收缩或者扩张,从而研究可能与该物种某些强/弱的生物学分子功能。
通过上述各实施例,可以基于三代测序技术针多样本量的比较转录组进行多种分析,归纳总结主要包括:
基因家族聚类,提取直系同源基因、旁系同源基因和物种特有基因;
宏观进化分析,构建系统发育树和分化时间分析;
ssr检测,功能标记开发;
选择压分析,挖掘研究材料不同亚种或近源物种快速进化基因;
物种特有基因数据库功能注释;
物种wgd事件检测;
个别家族的基因家族树分析。
下面对本发明实施例提供的基于三代测序技术针多样本量的比较转录组的分析进行举例说明。
例1、基于多样本量三代比较转录组分析方法应用8个样品宏观进化分析,其中,8个样品宏观进化分析,包括如下步骤:
a、pacbio下机数据质控,过滤接头,过滤片段低于300bp的reads。质控后,8个样品的cleandata数据如下表1所示:
表1
其中,samples为样品名,readsofinsert为roi序列的数目,meanreadlengthofinsert为roi序列质量值,meannumberofpasses为cell中所有zmw中序列的平均测序深度(passes)。
b、全长转录本的获取,根据软件smrtlink7.0,使用ccs程序针对cleandata数据提取高度一致的consensus序列,随后利用isoseq3中的cluster将一致性序列进行聚类,根据引物信息识别全长转录本和非全长转录本,然后利用polish程序结合非全长转录本校正全长转录本,最后根据过滤参数hp_min_accuracy(0.99)将全长转录本分为高质量全长转录本(>=0.99)和低质量全长转录本。最终获取高质量的全长转录本结果如下表2所示:
表2
其中,samples为样品名称,numberofpolishedhigh-qualityisoforms为高质量转录本总数,numberofpolishedlow-qualityisoforms为低质量转录本总数,total为总的转录本数目。
c、针对高质量全长转录本进行cds/pep预测。利用transdecoder-3.0.1对全长转录本进行orf预测,利用blast比对nr以及swissprot蛋白数据库,然后提取比对上的同源序列的位置识别orf。根据全长转录本预测的orf结果,选择最长的orf代表该基因。针对8个样品,一共预测的结果如下表3所示:
表3
d、基因家族聚类,利用blastp程序对pep序列进行all-vs-all比对,以e-value为1e-10进行搜索,选择besthitstop5%作为备选结果。用比特值代替e-value值,用最小二乘法将top5%的分数值拟合成一个线性模型,用rbnh方法将基因长度和系统距离标准化,确定同源组序列相似度的阈值为0.942,筛选出质量好的直系同源组图,通过归一化的比特值画出正交组的边缘连接,基于mcl,将直系同源正交组根据膨胀系数1.5进行聚类。基于聚类结果判断单拷贝直系同源基因、多拷贝旁系同源基因、物种特有基因以及物种共有基因,具体详细信息见下表4所示:
表4
其中,name表示物种名,onecopynum表示单拷贝同源,multicopynum表示多拷贝同源基因,unigenenum表示物种特有基因。
e、基于单拷贝直系同源基因构建物种系统发育树。分别针对1200个单拷贝基因家族的cds和pep序列进行多样本muscle全局比对,序列对齐后,剔除比对结果中gap区域超过50%的基因对,然后将所有单拷贝基因家族的比对结果进行串联,构建两个超大矩阵(分别是cds和pep序列),序列长度分别是氨基酸长度142277和cds长度426831,然后基于两个超大矩阵构建系统发育树,且两种构建系统发育树的方法结果一致。具体结果参看附图3,从结果来看,主要分为两个单系群,分别是f01、f02、f03、f08为一组;f04、f05、f06、f07为一组。图中节点上的数字代表bootstrap值。
f、根据构建的物种系统发育树,结合提供的化石时间,即根节点时间110.9myr,通过基因序列间的分歧程度以及序列的平均置换速率(分子钟)来估计速率恒定分支间的分歧时间,同时计算系统树上其它节点的发生时间,从而推测估算8个样品间的起源时间和分歧时间,具体结果见附图4,图中节点的数字是估算的分化时间,节点上的小矩形代表估算的分化的95%置信区间。
本发明实施例提供的一种基于三代测序检测多样本量比较转录组分析方法,有益效果包括:
(1)针对多样本量采用最佳互惠比对方式,运用比特值代替e-value值方法进行拟合并进行模型转换;(2)利用rbnh方法确定同源组序列相似度的阈值,从而筛选高质量的直系同源组图;(3)利用stag算法和stride算法构建有根物种树和有根基因树;(4)针对直系同源基因,采用三种鉴别方式进行选择压分析;(5)基于ks分布图的方式预测wgd事件;(6)开发出一套方法处理多样本量转录组数据。
在处理多样本量比较转录组数据时,针对检测的全长转录本进行cds/pep预测;随后进行宏观进化分析,主要包括系统发育分析和分化时间分析;针对预测的单拷贝直系同源基因进行ssr检测,标记开发;针对单拷贝直系同源基因进行选择压分析,挖掘物种适应性进化和快速进化基因;针对旁系同源基因个别家族进行基因家族树分析;针对特殊材料进行wgd事件分析。整体上,提供了一套完善的科学研究方法进行生物信息学的研究。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 一种预测PD潜在gene和m...
- 肠道微生物测序数据处理方法、...
- 肠道微生物测序数据处理方法、...
- 基于stacking集成的R...
- 一种基于蛋白质空间结构的二硫...
- 一种实验数据辅助的自适应策略...
- 成年人经口、皮肤暴露于双酚S...
- 一种模型预测的关键区域的分析...
- 一种高强度聚焦超声换能器的制...
- 一种肿瘤放射治疗中控制呼吸的...
- 还没有人留言评论。精彩留言会获得点赞!